Clear Sky Science · es
Eficiencia de genes y líneas celulares de CRISPR calculada mediante descomposición de tensores en cribados de deleción CRISPR-Cas9 a escala genómica
Encontrar las partes importantes de nuestra caja de herramientas del ADN
La edición genética con CRISPR se ha convertido en una de las herramientas más potentes de la biología moderna, permitiendo a los científicos desactivar miles de genes a la vez para ver cuáles son realmente importantes para la supervivencia celular. Pero convertir ese aluvión de datos experimentales en respuestas claras resulta sorprendentemente difícil. Este artículo presenta un enfoque matemático sencillo pero eficaz, llamado descomposición de tensores, que ayuda a los científicos a interpretar experimentos CRISPR masivos de forma más fiable, incluso cuando faltan algunas de las muestras de control habituales.
Por qué apagar genes es tan desordenado
En un experimento CRISPR a escala genómica típico, los investigadores usan muchas moléculas guía cortas, llamadas sgRNAs, para anular cada gen en diversas líneas celulares. En teoría, si la pérdida de un gen mata o debilita a las células, ese gen es esencial; si no ocurre gran cosa, probablemente sea no esencial. En la práctica, cada guía varía en su eficacia y distintos laboratorios miden los resultados de forma ligeramente diferente. Como resultado, los científicos deben combinar de algún modo las lecturas ruidosas de muchas guías, genes y líneas celulares en una única puntuación que indique cuán importante es realmente cada gen. Muchos métodos actuales hacen esto con modelos estadísticos elaborados y a menudo requieren muestras de control especiales como anclas.
Una forma simple de mirar los datos en varias direcciones a la vez
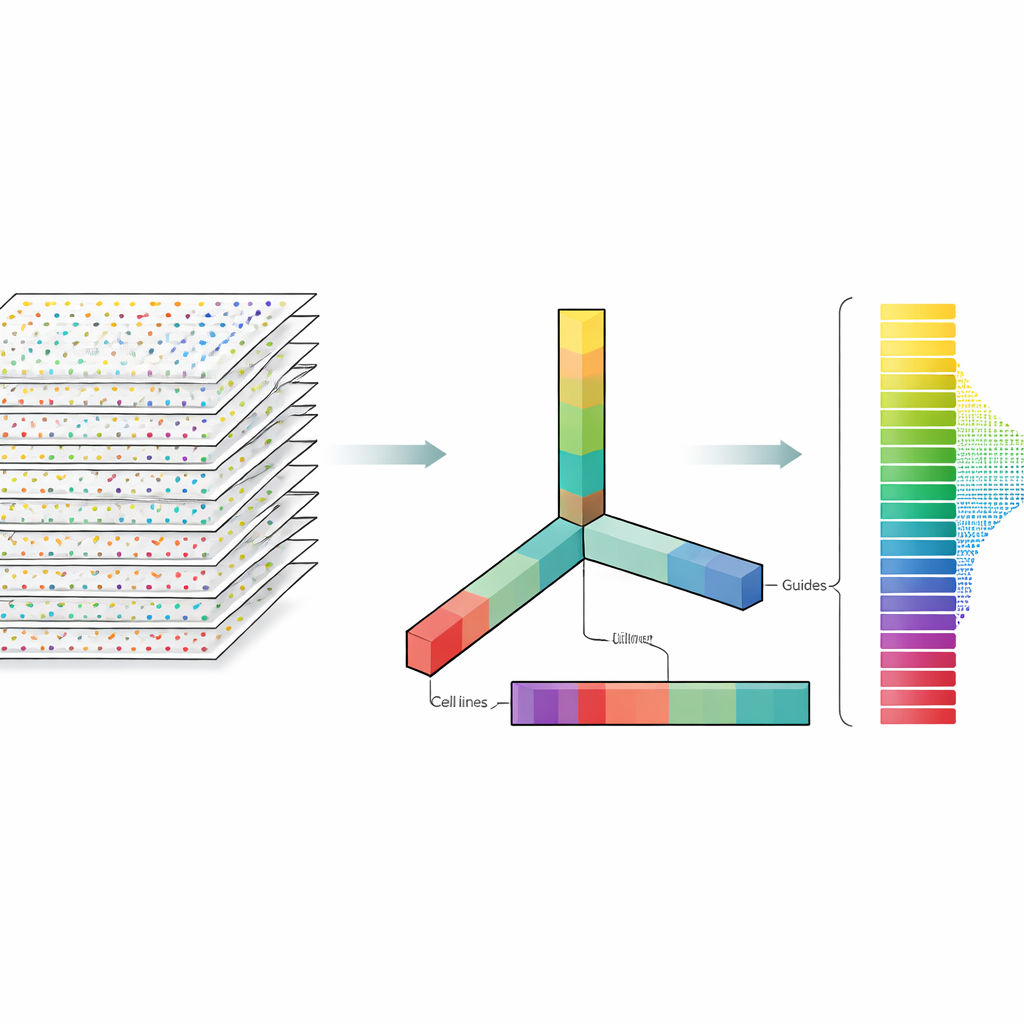
Los autores tratan los datos de CRISPR no como una hoja de cálculo plana sino como un bloque multidimensional que se puede cortar en varias direcciones a la vez: genes, guías, líneas celulares y réplicas experimentales. La descomposición de tensores es una técnica de álgebra lineal que descompone este bloque en un conjunto de patrones básicos más pesos que indican con qué intensidad aparece cada patrón. Sin necesidad de etiquetas previas ni entrenamiento, estos patrones pueden resaltar qué genes se comportan como genes esenciales conocidos y qué líneas celulares comparten patrones de respuesta similares. De forma crucial, el método integra tanto las múltiples guías por gen como los múltiples perfiles experimentales desde el inicio, en lugar de analizar cada archivo por separado y fusionar resultados más tarde.
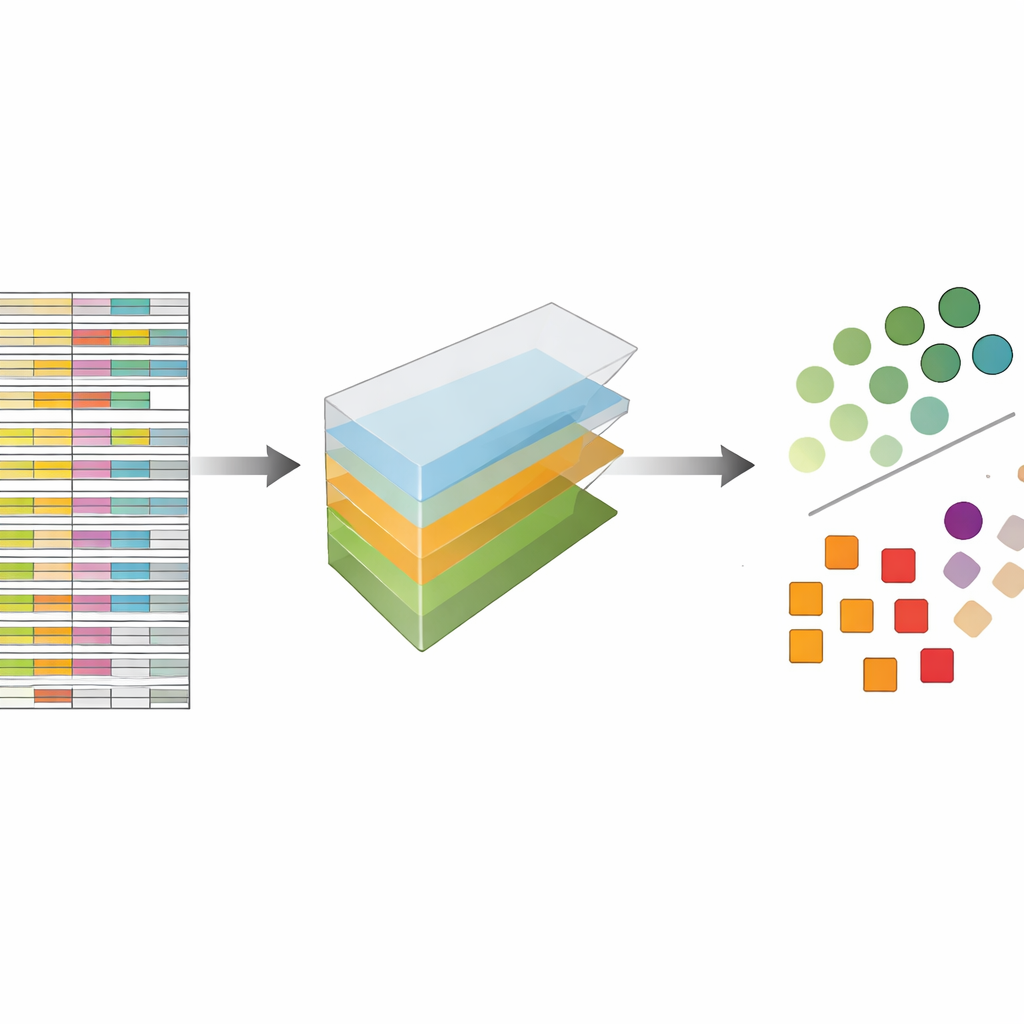
Igualando a los mejores métodos modernos sin adornos innecesarios
Para evaluar la eficacia de este enfoque, los autores lo aplicaron a cinco colecciones grandes y ampliamente usadas de cribados CRISPR que previamente habían sido analizadas con un método líder llamado JACKS, así como con otras herramientas de vanguardia. Plantearon una pregunta simple: ¿qué tan bien puede su método distinguir una lista publicada de genes esenciales de genes no esenciales? Medido por una puntuación de precisión estándar conocida como área bajo la curva, la descomposición de tensores rindió a la par con JACKS en los conjuntos de datos, alcanzando a menudo valores alrededor de 0,8, lo que se considera sólido en este contexto. Aún más llamativo, las formas detalladas de las curvas de rendimiento coincidieron estrechamente con las de JACKS, lo que sugiere que el método más simple está capturando gran parte de la misma señal biológica que el enfoque bayesiano más complejo.
Funciona cuando faltan controles y los números se mantienen crudos
Algunos de los conjuntos de datos carecían de las muestras de control habituales en las que muchos métodos se apoyan, y aun así la descomposición de tensores funcionó bien. En conjuntos de datos con controles, el método seleccionó de forma natural patrones que separaban las muestras de control de las tratadas, lo que le ayudó a identificar genes esenciales. En conjuntos sin controles, en su lugar descubrió patrones que seguían de cerca medidas independientes de cuán eficientemente funciona CRISPR en cada línea celular, extraídas de un gran proyecto sobre dependencias en cáncer. Otra sorpresa práctica fue que el método rindió igual de bien al usar los recuentos brutos que al usar transformaciones logarítmicas, un paso de preprocesado común pero no siempre justificado. Este hallazgo abre la posibilidad de que los cribados CRISPR no requieran tanto ajuste numérico como a menudo se asume.
Qué significa esto para estudios futuros de edición génica
En conjunto, el estudio muestra que una lente matemática relativamente simple puede compararse con modelos sofisticados y muy ajustados para analizar cribados CRISPR a gran escala. Al manejar conjuntamente muchas guías y muchos experimentos a la vez, la descomposición de tensores puede separar de forma fiable genes esenciales de no esenciales y revelar diferencias en la eficacia de CRISPR entre líneas celulares, incluso sin controles ideales. Para los no especialistas, el mensaje clave es que formas más inteligentes de mirar los mismos datos pueden hacer que los experimentos de edición génica sean más fiables y fáciles de comparar, ayudando a los investigadores a identificar más rápidamente los genes que importan para la salud y la enfermedad.
Cita: Taguchi, YH., Turki, T. Gene and cell line efficiency of CRISPR computed by tensor decomposition in genome-wide CRISPR-Cas9 knockout screens. Sci Rep 16, 13605 (2026). https://doi.org/10.1038/s41598-026-43209-0
Palabras clave: cribados CRISPR, esencialidad génica, descomposición de tensores, eficiencia de sgRNA, líneas celulares de cáncer