Clear Sky Science · fr
Efficacité des gènes et des lignées cellulaires du CRISPR calculée par décomposition de tenseur dans des criblages CRISPR-Cas9 de l’ensemble du génome
Identifier les parties importantes de notre boîte à outils génétique
L’édition génétique CRISPR est devenue l’un des outils les plus puissants de la biologie moderne, permettant aux chercheurs d’éteindre des milliers de gènes à la fois pour déterminer lesquels sont réellement essentiels à la survie cellulaire. Mais transformer cet afflux de données expérimentales en réponses claires est étonnamment difficile. Cet article présente une approche mathématique simple mais efficace, appelée décomposition de tenseur, qui aide les scientifiques à interpréter ces vastes expériences CRISPR de façon plus fiable, même lorsque certains échantillons de contrôle habituels font défaut.
Pourquoi le fait d’inactiver des gènes est si compliqué
Dans un criblage CRISPR à l’échelle du génome classique, les chercheurs utilisent de nombreuses courtes molécules guides, appelées sgRNA, pour inactiver chaque gène dans de nombreuses lignées cellulaires différentes. En théorie, si la perte d’un gène tue ou affaiblit les cellules, ce gène est essentiel ; si rien de notable ne se produit, il est probablement non essentiel. En pratique, chaque guide varie dans son efficacité, et différents laboratoires mesurent les résultats de façons légèrement différentes. Par conséquent, il faut combiner d’une manière ou d’une autre les mesures bruitées provenant de nombreux guides, gènes et lignées cellulaires en un score unique qui reflète l’importance réelle de chaque gène. De nombreuses méthodes actuelles utilisent des modèles statistiques élaborés et exigent souvent des échantillons de contrôle spécifiques comme points d’ancrage.
Une manière simple de regarder les données dans plusieurs directions à la fois
Les auteurs considèrent les données CRISPR non pas comme un tableau plat mais comme un bloc multidimensionnel qu’on peut découper selon plusieurs axes à la fois : gènes, guides, lignées cellulaires et répétitions expérimentales. La décomposition de tenseur est une technique d’algèbre linéaire qui décompose ce bloc en un ensemble de motifs de base plus des poids indiquant l’importance de chaque motif. Sans nécessité d’étiquettes préalables ni d’apprentissage supervisé, ces motifs peuvent mettre en évidence quels gènes se comportent comme des gènes essentiels connus et quelles lignées cellulaires partagent des schémas de réponse similaires. De façon cruciale, la méthode intègre dès le départ à la fois les multiples guides par gène et les multiples profils expérimentaux, au lieu d’analyser chaque fichier séparément puis de fusionner les résultats a posteriori.
Comparer aux meilleures méthodes modernes sans artifices
Pour évaluer l’efficacité de l’approche, les auteurs l’ont appliquée à cinq grandes collections de criblages CRISPR largement utilisées, qui avaient été précédemment analysées avec une méthode de référence appelée JACKS ainsi qu’avec d’autres outils de pointe. Ils ont posé une question simple : dans quelle mesure leur méthode peut-elle distinguer une liste publiée de gènes essentiels de gènes non essentiels ? Mesurée par un score d’exactitude standard connu sous le nom d’aire sous la courbe, la décomposition de tenseur a obtenu des performances comparables à JACKS selon les jeux de données, atteignant souvent des valeurs autour de 0,8, ce qui est considéré comme solide dans ce contexte. Plus frappant encore, les formes détaillées des courbes de performance correspondaient étroitement à celles de JACKS, suggérant que la méthode plus simple capte une grande partie du même signal biologique que l’approche bayésienne plus complexe.
Fonctionne lorsque les contrôles manquent et avec les nombres bruts
Certains des ensembles de données étaient dépourvus des échantillons de contrôle habituels sur lesquels reposent de nombreuses méthodes, et pourtant la décomposition de tenseur a continué à bien fonctionner. Dans les jeux de données contenant des contrôles, la méthode a naturellement isolé des motifs séparant les échantillons témoins des échantillons traités, ce qui l’aidait à repérer les gènes essentiels. Dans les jeux de données sans contrôles, elle a découvert des motifs qui suivaient de près des mesures indépendantes de l’efficacité du CRISPR dans chaque lignée cellulaire, tirées d’un vaste projet sur les dépendances des cancers. Autre surprise pratique : la méthode a donné des résultats tout aussi bons en utilisant les comptes bruts qu’en utilisant des transformations logarithmiques, une étape de prétraitement courante mais pas toujours justifiée. Cette observation suggère que les criblages CRISPR pourraient nécessiter moins de retouches numériques qu’on ne le suppose souvent.
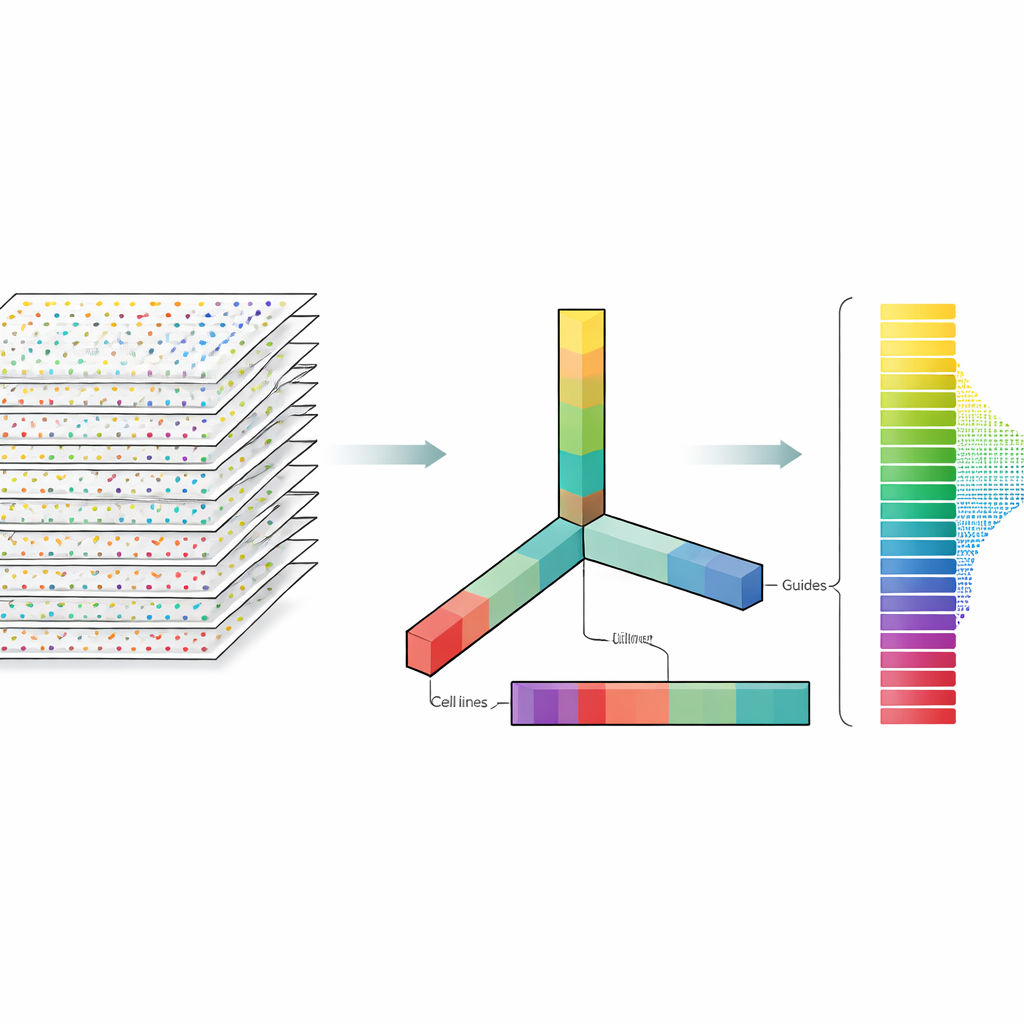
Ce que cela implique pour les futures études d’édition génétique
Globalement, l’étude montre qu’une lentille mathématique relativement simple peut rivaliser avec des modèles sophistiqués et finement réglés pour l’analyse de criblages CRISPR à grande échelle. En traitant conjointement de nombreux guides et de nombreuses expériences, la décomposition de tenseur peut séparer de manière fiable les gènes essentiels des non essentiels et révéler des différences dans l’efficacité du CRISPR selon les lignées cellulaires, même sans contrôles idéaux. Pour les non-spécialistes, le message clé est que de meilleures façons d’examiner les mêmes données peuvent rendre les expériences d’édition génique plus fiables et plus faciles à comparer, aidant les chercheurs à identifier plus rapidement les gènes qui comptent le plus pour la santé et la maladie.
Citation: Taguchi, YH., Turki, T. Gene and cell line efficiency of CRISPR computed by tensor decomposition in genome-wide CRISPR-Cas9 knockout screens. Sci Rep 16, 13605 (2026). https://doi.org/10.1038/s41598-026-43209-0
Mots-clés: criblages CRISPR, essentiels génétiques, décomposition de tenseur, efficacité des sgRNA, lignées cellulaires cancéreuses