Clear Sky Science · de
Gen- und Zelllinien-Effizienz von CRISPR berechnet durch Tensorzerlegung in genomweiten CRISPR-Cas9-Knockout-Screens
Die wichtigen Teile unseres DNA-Werkzeugs finden
Die CRISPR-Geneditierung ist eines der mächtigsten Werkzeuge der modernen Biologie geworden: Sie erlaubt es Wissenschaftlern, tausende Gene gleichzeitig auszuschalten, um zu sehen, welche davon für das Überleben von Zellen wirklich wichtig sind. Aber aus dieser Flut experimenteller Daten klare Antworten zu gewinnen, ist überraschend schwierig. Diese Arbeit stellt einen einfachen, aber wirkungsvollen mathematischen Ansatz vor, die sogenannte Tensorzerlegung, die Forschern hilft, massive CRISPR-Experimente zuverlässiger zu interpretieren — selbst wenn einige der üblichen Kontrollproben fehlen.
Warum Gene auszuschalten so unordentlich ist
In einem typischen genomweiten CRISPR-Experiment verwenden Forschende viele kurze Führungsmoleküle, sogenannte sgRNAs, um jedes Gen in vielen verschiedenen Zelllinien auszuschalten. Theoretisch gilt: Wenn das Fehlen eines Gens Zellen tötet oder schwächt, ist dieses Gen essentiell; wenn kaum etwas passiert, ist es vermutlich nicht essentiell. In der Praxis variiert jedoch jede Führungssequenz in ihrer Wirksamkeit, und verschiedene Labore messen Ergebnisse leicht unterschiedlich. Folglich müssen Wissenschaftler die verrauschten Messwerte vieler Führungen, Gene und Zelllinien zu einer einzigen Bewertung zusammenführen, die aussagt, wie wichtig jedes Gen wirklich ist. Viele aktuelle Methoden tun dies mit aufwändigen statistischen Modellen und benötigen häufig spezielle Kontrollproben als Bezugspunkte.
Eine einfache Art, Daten gleichzeitig in mehrere Richtungen zu betrachten
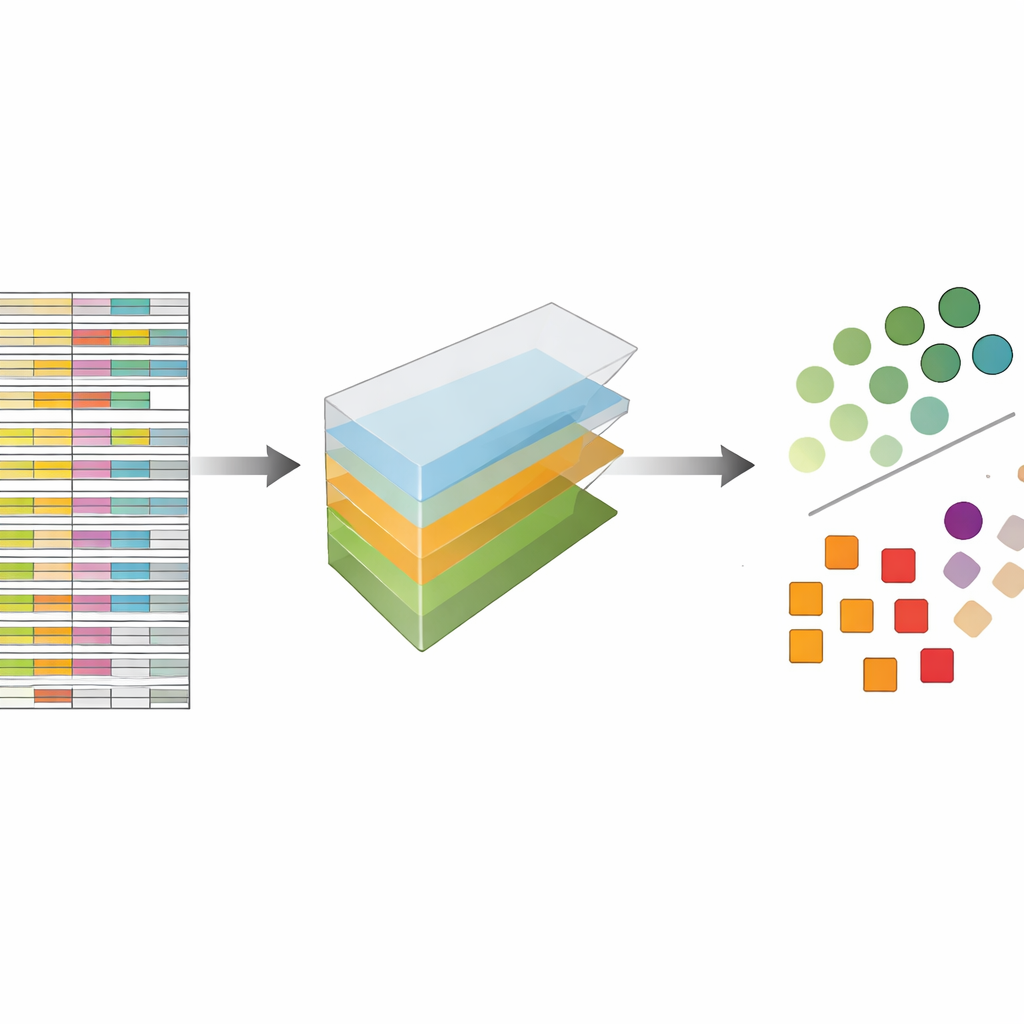
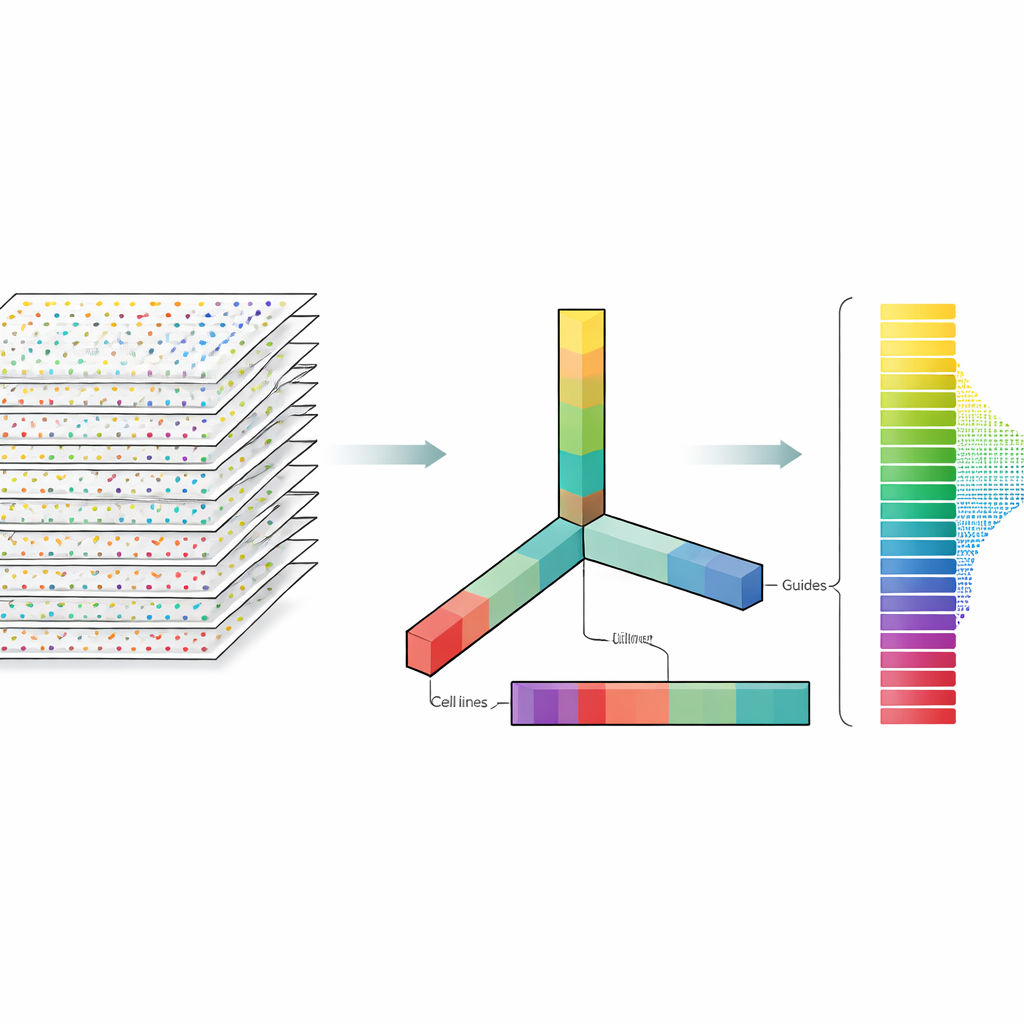
Die Autorinnen und Autoren betrachten CRISPR-Daten nicht als flache Tabelle, sondern als mehrdimensionalen Block, den sie in mehreren Richtungen gleichzeitig aufschneiden können: Gene, Führungen, Zelllinien und experimentelle Wiederholungen. Die Tensorzerlegung ist eine Technik der linearen Algebra, die diesen Block in eine Menge grundlegender Muster zerlegt sowie Gewichtungen liefert, die angeben, wie stark jedes Muster auftritt. Ohne vorherige Labels oder Training können diese Muster hervorheben, welche Gene sich wie bekannte essentielle Gene verhalten und welche Zelllinien ähnliche Reaktionsmuster zeigen. Entscheidend ist, dass die Methode sowohl mehrere Führungen pro Gen als auch mehrere experimentelle Profile von Anfang an integriert, statt jede Datei separat zu analysieren und die Ergebnisse später zusammenzuführen.
So gut wie die besten modernen Methoden — ohne Schnickschnack
Um zu testen, wie gut dieser Ansatz funktioniert, wendeten die Autorinnen und Autoren ihn auf fünf große, weit verbreitete CRISPR-Screening-Sammlungen an, die zuvor mit einer führenden Methode namens JACKS sowie mit anderen modernen Werkzeugen analysiert worden waren. Sie stellten eine einfache Frage: Wie gut kann ihre Methode eine veröffentlichte Liste essentieller Gene von nicht-essenziellen unterscheiden? Gemessen an einer gebräuchlichen Genauigkeitsmetrik, der Fläche unter der Kurve, erzielte die Tensorzerlegung vergleichbare Werte wie JACKS über die Datensätze hinweg und erreichte oft Werte um 0,8, was in diesem Kontext als stark gilt. Noch auffälliger war, dass die detaillierten Formen der Leistungskurven JACKS eng ähnelten, was darauf hindeutet, dass die einfachere Methode einen Großteil desselben biologischen Signals erfasst wie der komplexere bayesianische Ansatz.
Funktioniert, wenn Kontrollen fehlen und Zahlen roh bleiben
Einige der Datensätze hatten nicht die üblichen Kontrollproben, auf die viele Methoden angewiesen sind, doch die Tensorzerlegung funktionierte dennoch gut. In Datensätzen mit Kontrollen wählte die Methode auf natürliche Weise Muster aus, die Kontrollproben von behandelten Proben trennten, was ihr half, essentielle Gene zu finden. In Datensätzen ohne Kontrollen entdeckte sie stattdessen Muster, die eng mit unabhängigen Messungen der CRISPR-Effizienz in jeder Zelllinie übereinstimmten, gewonnen aus einem großen Krebs-Abhängigkeitsprojekt. Eine weitere praktische Überraschung war, dass die Methode mit den rohen Zähldaten genauso gute Ergebnisse lieferte wie mit logarithmischen Transformationen — einem gebräuchlichen, aber nicht immer gerechtfertigten Vorverarbeitungsschritt. Dieses Ergebnis legt nahe, dass CRISPR-Screens möglicherweise nicht so viele numerische Anpassungen benötigen, wie oft angenommen wird.
Was das für zukünftige Geneditier-Studien bedeutet
Insgesamt zeigt die Studie, dass eine vergleichsweise einfache mathematische Perspektive mit ausgefeilten, stark abgestimmten Modellen zur Analyse groß angelegter CRISPR-Screens mithalten kann. Indem sie viele Führungen und viele Experimente zugleich betrachtet, kann die Tensorzerlegung zuverlässig essentielle von nicht-essenziellen Genen trennen und Unterschiede in der CRISPR-Wirksamkeit zwischen Zelllinien aufdecken — selbst ohne ideale Kontrollen. Für Nicht-Spezialisten lautet die Kernbotschaft: Schlauere Sichtweisen auf die gleichen Daten können Geneditier-Experimente vertrauenswürdiger und leichter vergleichbar machen und Forschern helfen, schneller die Gene zu identifizieren, die für Gesundheit und Krankheit am wichtigsten sind.
Zitation: Taguchi, YH., Turki, T. Gene and cell line efficiency of CRISPR computed by tensor decomposition in genome-wide CRISPR-Cas9 knockout screens. Sci Rep 16, 13605 (2026). https://doi.org/10.1038/s41598-026-43209-0
Schlüsselwörter: CRISPR-Screens, Gen-Essentialität, Tensorzerlegung, sgRNA-Effizienz, Krebszelllinien