Clear Sky Science · pt
Eficiência de genes e linhagens celulares do CRISPR calculada por decomposição de tensores em triagens genômicas CRISPR-Cas9 de nocaute
Encontrando as Partes Importantes do Nosso Kit Genético
A edição gênica por CRISPR tornou-se uma das ferramentas mais poderosas da biologia moderna, permitindo que cientistas desativem milhares de genes de uma vez para ver quais realmente importam para a sobrevivência celular. Mas transformar esse dilúvio de dados experimentais em respostas claras é surpreendentemente difícil. Este artigo apresenta uma abordagem matemática simples porém eficaz, chamada decomposição de tensores, que ajuda os pesquisadores a interpretar esses experimentos massivos de CRISPR de forma mais confiável, mesmo quando algumas das amostras de controle habituais estão ausentes.
Por que Desligar Genes É Tão Complicado
Em um experimento típico de CRISPR em escala genômica, os pesquisadores usam muitas moléculas guia curtas, chamadas sgRNAs, para nocautear cada gene em várias linhagens celulares diferentes. Em teoria, se perder um gene mata ou enfraquece as células, esse gene é essencial; se nada de importante acontece, provavelmente é não essencial. Na prática, cada guia varia em quão bem funciona, e diferentes laboratórios medem os resultados de maneiras ligeiramente diferentes. Como resultado, os cientistas precisam combinar de alguma forma as leituras ruidosas de muitas guias, genes e linhagens celulares em uma única pontuação que diga o quão importante cada gene realmente é. Muitos métodos atuais fazem isso com modelos estatísticos elaborados e frequentemente exigem amostras de controle especiais como âncoras.
Uma Maneira Simples de Olhar os Dados em Várias Direções ao Mesmo Tempo
Os autores tratam os dados de CRISPR não como uma planilha plana, mas como um bloco multidimensional que pode ser fatiado em várias direções ao mesmo tempo: genes, guias, linhagens celulares e repetições experimentais. A decomposição de tensores é uma técnica de álgebra linear que divide esse bloco em um conjunto de padrões básicos mais pesos que indicam quão fortemente cada padrão aparece. Sem necessidade de rótulos prévios ou treinamento, esses padrões podem destacar quais genes se comportam como genes essenciais conhecidos e quais linhagens celulares compartilham padrões de resposta semelhantes. Crucialmente, o método integra tanto múltiplas guias por gene quanto múltiplos perfis experimentais desde o início, em vez de analisar cada arquivo separadamente e mesclar os resultados depois.
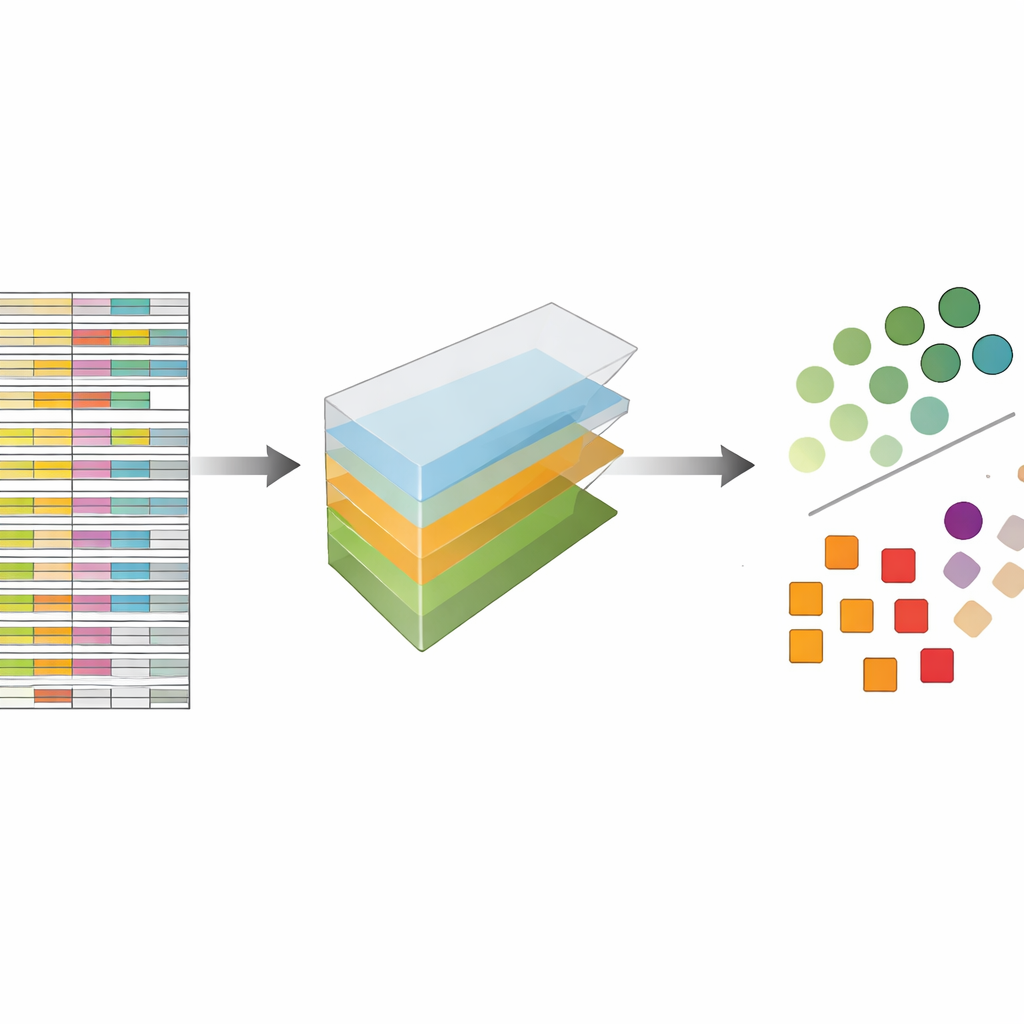
Desempenho Igual aos Melhores Métodos Modernos sem Recursos Extras
Para testar quão bem essa abordagem funciona, os autores a aplicaram a cinco grandes coleções de triagens CRISPR amplamente usadas que haviam sido analisadas anteriormente com um método de referência chamado JACKS, assim como com outras ferramentas de ponta. Eles fizeram uma pergunta simples: quão bem seu método consegue distinguir uma lista publicada de genes essenciais de genes não essenciais? Medido por uma pontuação de acurácia padrão conhecida como área sob a curva, a decomposição de tensores teve desempenho comparável ao JACKS entre os conjuntos de dados, frequentemente alcançando valores em torno de 0,8, o que é considerado forte nesse contexto. Ainda mais notável, as formas detalhadas das curvas de desempenho bateram de perto com as do JACKS, sugerindo que o método mais simples está capturando grande parte do mesmo sinal biológico que a abordagem bayesiana mais complexa.
Funciona Quando Faltam Controles e os Números Permanecem Brutos
Alguns dos conjuntos de dados não tinham as amostras de controle usuais das quais muitos métodos dependem, mas a decomposição de tensores ainda funcionou bem. Em conjuntos de dados com controles, o método naturalmente destacou padrões que separavam amostras de controle das tratadas, o que ajudou a identificar genes essenciais. Em conjuntos sem controles, ele descobriu padrões que acompanharam de perto medidas independentes de quão eficientemente o CRISPR funciona em cada linhagem celular, extraídas de um grande projeto de dependência em câncer. Outra surpresa prática foi que o método teve desempenho equivalente ao usar os contagens brutas ou transformações logarítmicas, um passo de pré-processamento comum, mas nem sempre justificado. Essa constatação sugere que as triagens CRISPR podem não precisar de tanta manipulação numérica quanto frequentemente se assume.
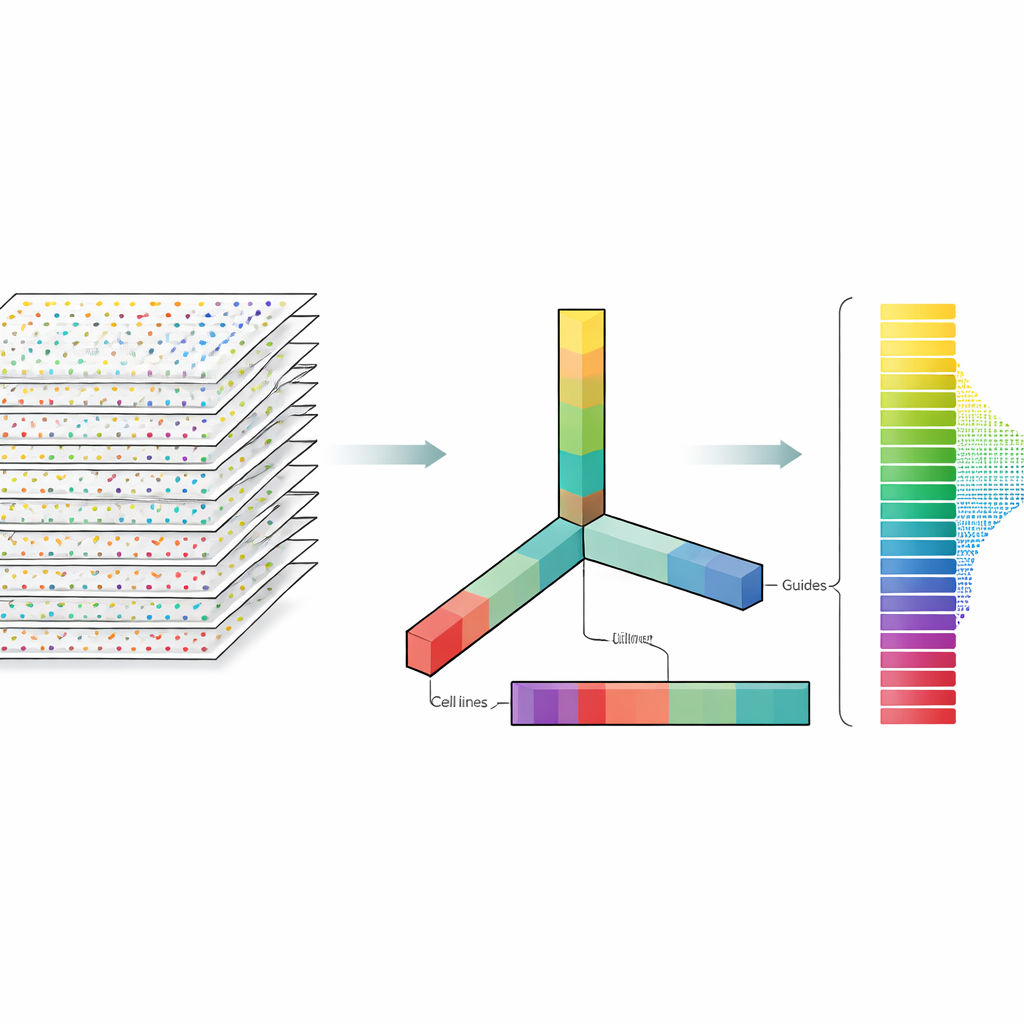
O Que Isso Significa para Estudos Futuros de Edição Gênica
No geral, o estudo mostra que uma lente matemática relativamente simples pode competir com modelos sofisticados e altamente ajustados para analisar triagens CRISPR em larga escala. Ao lidar conjuntamente com muitas guias e muitos experimentos ao mesmo tempo, a decomposição de tensores pode separar de forma confiável genes essenciais de não essenciais e revelar diferenças em quão bem o CRISPR funciona entre linhagens celulares, mesmo sem controles ideais. Para não especialistas, a mensagem-chave é que maneiras mais inteligentes de olhar os mesmos dados podem tornar experimentos de edição gênica mais confiáveis e fáceis de comparar, ajudando os pesquisadores a identificar mais rapidamente os genes que mais importam para a saúde e a doença.
Citação: Taguchi, YH., Turki, T. Gene and cell line efficiency of CRISPR computed by tensor decomposition in genome-wide CRISPR-Cas9 knockout screens. Sci Rep 16, 13605 (2026). https://doi.org/10.1038/s41598-026-43209-0
Palavras-chave: triagens CRISPR, essencialidade gênica, decomposição de tensores, eficiência de sgRNA, linhagens celulares de câncer