Clear Sky Science · pl
Wydajność genów i linii komórkowych CRISPR obliczona przez dekompozycję tensora w przesiewach CRISPR-Cas9 obejmujących cały genom
Odnajdywanie ważnych elementów naszego zestawu narzędzi DNA
Edytowanie genów metodą CRISPR stało się jednym z najsilniejszych narzędzi współczesnej biologii, pozwalając naukowcom wyłączać tysiące genów jednocześnie, by sprawdzić, które z nich rzeczywiście mają znaczenie dla przeżycia komórek. Przekształcenie tego potoku danych eksperymentalnych w jasne odpowiedzi jest jednak zaskakująco trudne. W artykule przedstawiono proste, ale skuteczne podejście matematyczne, zwane dekompozycją tensora, które pomaga badaczom odczytywać ogromne eksperymenty CRISPR bardziej niezawodnie, nawet gdy brakuje niektórych zwykłych próbek kontrolnych.
Dlaczego wyłączanie genów jest takie nieporządne
W typowym eksperymencie CRISPR obejmującym cały genom badacze używają wielu krótkich molekuł prowadzących, zwanych sgRNA, aby wybić każdy gen w wielu różnych liniach komórkowych. W teorii, jeśli utrata genu zabija lub osłabia komórki, gen jest niezbędny; jeśli niewiele się dzieje, prawdopodobnie nie jest istotny. W praktyce każdy prowadnik różni się skutecznością, a różne laboratoria mierzą wyniki nieco inaczej. W efekcie naukowcy muszą w jakiś sposób połączyć hałaśliwe odczyty z wielu prowadników, genów i linii komórkowych w pojedynczy wynik, który mówi, jak ważny jest dany gen. Wiele obecnych metod robi to przy użyciu rozbudowanych modeli statystycznych i często wymaga specjalnych próbek kontrolnych jako punktów odniesienia.
Prosty sposób patrzenia na dane w kilku wymiarach naraz
Autorzy traktują dane CRISPR nie jako płaski arkusz kalkulacyjny, lecz jako wielowymiarowy blok, który można kroić w kilku kierunkach jednocześnie: geny, prowadniki, linie komórkowe i powtórzenia eksperymentalne. Dekompozycja tensora to technika algebry liniowej, która rozbija ten blok na zestaw podstawowych wzorców oraz wag określających, jak silnie każdy wzorzec występuje. Bez potrzeby wcześniejszych etykiet czy trenowania, te wzorce mogą uwypuklić, które geny zachowują się jak znane geny niezbędne oraz które linie komórkowe wykazują podobne wzory odpowiedzi. Co kluczowe, metoda integruje zarówno wiele prowadników na gen, jak i wiele profili eksperymentalnych od samego początku, zamiast analizować każdy plik oddzielnie i łączyć wyniki później.
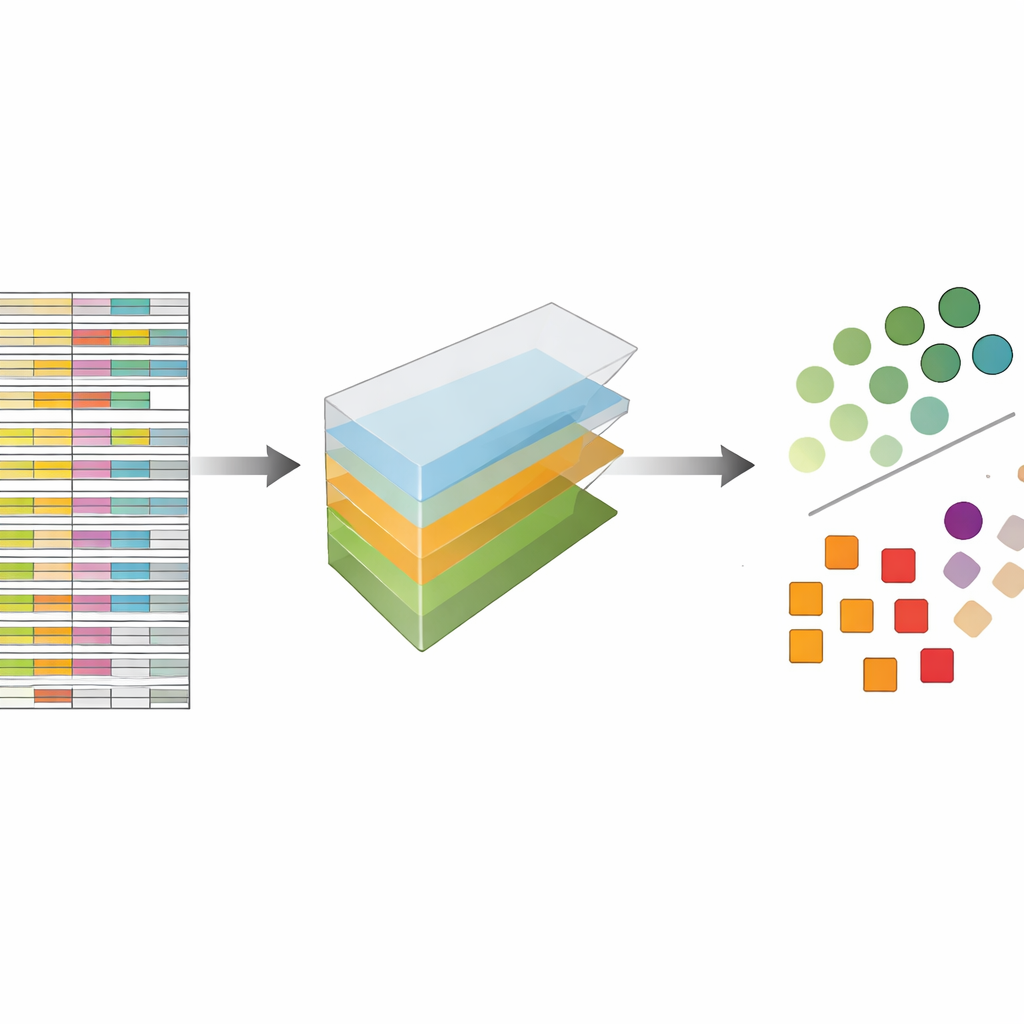
Porównywalna z najlepszymi współczesnymi metodami bez dodatkowych bajerów
Aby sprawdzić, jak dobrze to podejście działa, autorzy zastosowali je do pięciu dużych, powszechnie używanych kolekcji przesiewów CRISPR, które wcześniej analizowano wiodącą metodą o nazwie JACKS oraz innymi narzędziami nowej generacji. Postawili proste pytanie: jak dobrze ich metoda potrafi odróżnić opublikowaną listę genów niezbędnych od genów nieistotnych? Mierzona standardowym wskaźnikiem dokładności znanym jako pole pod krzywą, dekompozycja tensora wypadła równie dobrze jak JACKS w różnych zestawach danych, często osiągając wartości około 0,8, co w tym kontekście uważa się za silny wynik. Co jeszcze bardziej uderzające, kształty krzywych wydajności ściśle odpowiadały tym z JACKS, co sugeruje, że prostsza metoda wyłapuje dużą część tego samego sygnału biologicznego co bardziej złożone podejście bayesowskie.
Działa gdy brakuje kontroli i gdy liczby są surowe
Niektóre z zestawów danych nie miały zwykłych próbek kontrolnych, na których wiele metod polega, a mimo to dekompozycja tensora wciąż dobrze sobie radziła. W zestawach z kontrolami metoda naturalnie wyodrębniała wzorce oddzielające próbki kontrolne od poddanych zabiegowi, co pomagało w wykrywaniu genów niezbędnych. W zestawach bez kontroli odkrywała natomiast wzorce ściśle korelujące z niezależnymi miarami efektywności działania CRISPR w każdej linii komórkowej, pochodzącymi z dużego projektu badającego zależności nowotworowe. Kolejnym praktycznym zaskoczeniem było to, że metoda radziła sobie równie dobrze, używając surowych danych liczebnościowych, jak i danych po logarytmicznej transformacji — powszechnym, lecz nie zawsze uzasadnionym kroku wstępnego. To odkrycie sugeruje, że przesiewy CRISPR mogą nie wymagać tak intensywnej obróbki liczb, jak się zwykle zakłada.
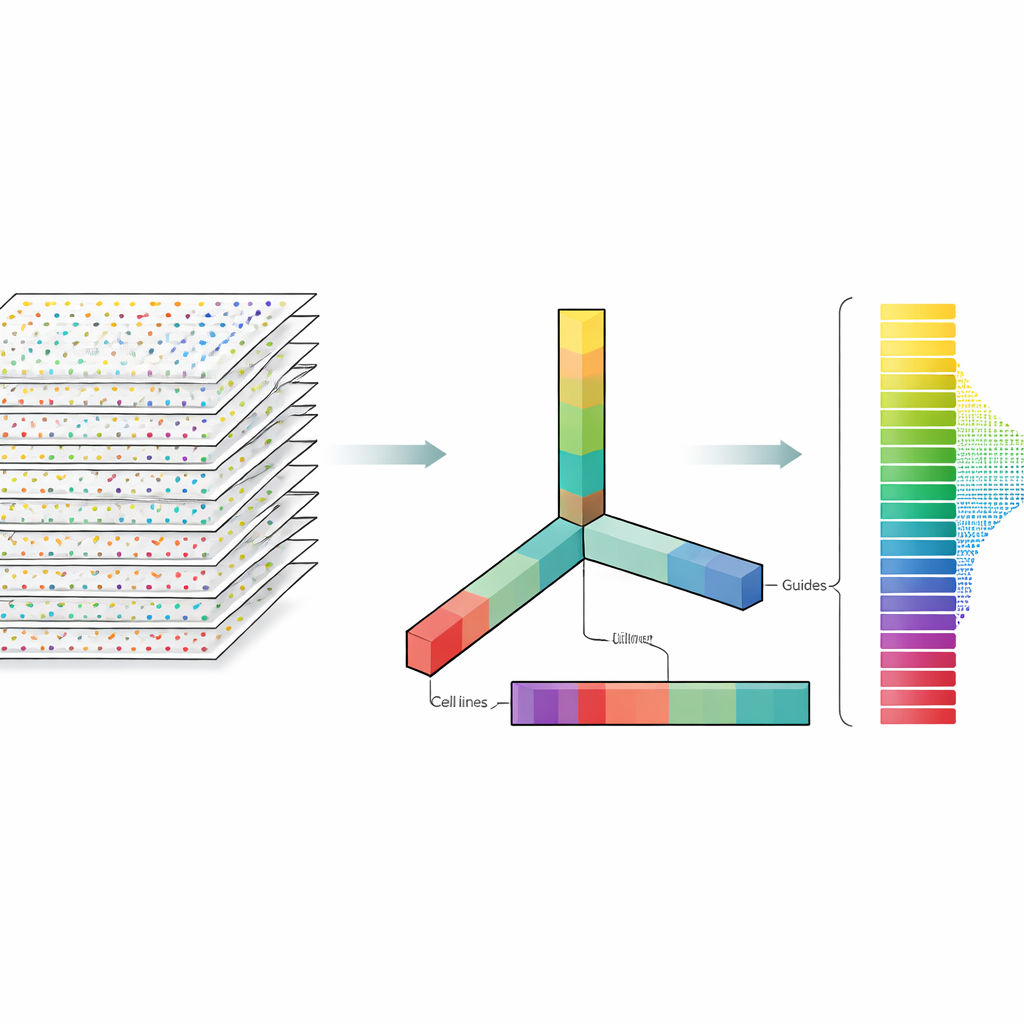
Co to oznacza dla przyszłych badań nad edycją genów
Podsumowując, badanie pokazuje, że stosunkowo prosta matematyczna soczewka może stać ramię w ramię z wysublimowanymi, silnie strojonych modelami do analizy przesiewów CRISPR na dużą skalę. Poprzez jednoczesne uwzględnienie wielu prowadników i wielu eksperymentów, dekompozycja tensora może niezawodnie oddzielać geny niezbędne od nieistotnych i ujawniać różnice w skuteczności CRISPR między liniami komórkowymi, nawet bez idealnych kontroli. Dla osób niebędących specjalistami kluczowy przekaz jest taki, że mądrzejsze sposoby patrzenia na te same dane mogą uczynić eksperymenty z edycją genów bardziej wiarygodnymi i łatwiejszymi do porównania, pomagając badaczom szybciej zidentyfikować geny mające największe znaczenie dla zdrowia i chorób.
Cytowanie: Taguchi, YH., Turki, T. Gene and cell line efficiency of CRISPR computed by tensor decomposition in genome-wide CRISPR-Cas9 knockout screens. Sci Rep 16, 13605 (2026). https://doi.org/10.1038/s41598-026-43209-0
Słowa kluczowe: przesiewy CRISPR, niezbędność genów, dekompozycja tensora, wydajność sgRNA, linie komórek nowotworowych