Clear Sky Science · sv
Ökad känslighet för 4-HNE‑inducerad toxicitet och försämrad utveckling i en modell för ALDH4A1‑bristig pediatrisk epilepsi med varianten S352L
När cellens städbesättning fallerar
Vissa barn utvecklar svårbehandlad epilepsi och livslånga inlärningssvårigheter av orsaker som fortfarande är delvis dunkla. Denna studie undersöker en sådan orsak: sällsynta förändringar i ett enstaka enzym inne i cellernas kraftverk, mitokondrierna. Genom att kartlägga hur detta enzym normalt rensar toxiska biprodukter och vad som händer när det sviktar, ger forskarna nya ledtrådar till varför anfall uppstår, varför hjärnan kan få nedsatt utveckling och hur vi skulle kunna utforma bättre behandlingar.
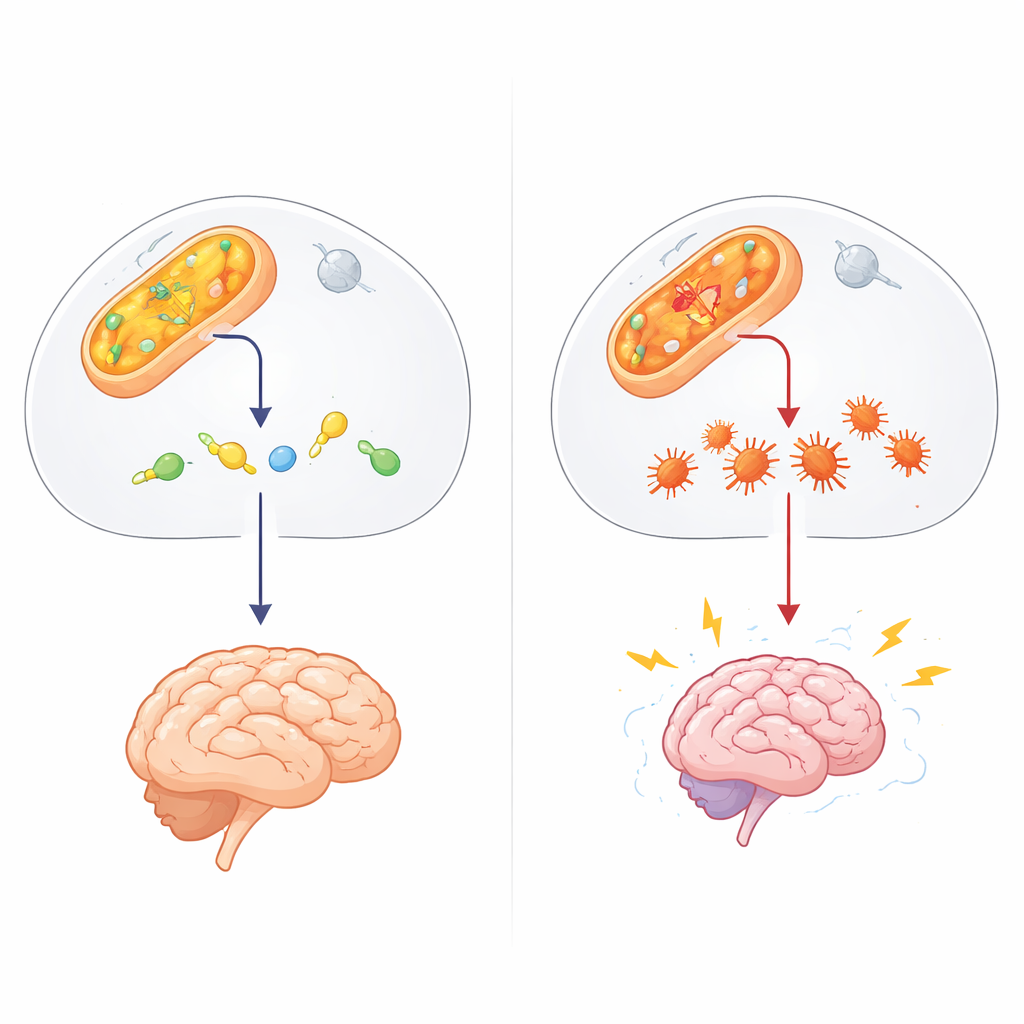
En dold väktare i cellkemin
Arbetet kretsar kring ALDH4A1, en del av en stor enzymfamilj som avgiftar aldehyder — mycket reaktiva kemiska rester som bildas vid normal ämnesomsättning, miljöpåverkan och vissa läkemedel. ALDH4A1 bryter normalt ner en förening kopplad till aminosyran prolin och hjälper till att hålla vitamin B6 aktivt. När ALDH4A1 saknas eller är defekt utvecklar människor Hyperprolinemi typ II, en sällsynt barnsjukdom med mycket höga prolinhalter, anfall och utvecklingsförsening. Hittills har de flesta förklaringar pekat på överskott av prolin och brist på vitamin B6. Dessa idéer kan dock inte fullt ut förklara patienter som inte förbättras av vitamintillskott eller de förbryllande skillnaderna mellan närliggande sjukdomar.
En toxisk molekyl i rampljuset
Författarna misstänkte en annan aktör: 4‑hydroxynonenal (4‑HNE), en klibbig aldehyd som bildas när fetter i cellmembran skadas av oxidativ stress, till exempel under anfall. 4‑HNE kan binda sig till proteiner och DNA, vilket får dem att felveckas och klumpa ihop sig. I provrörsexperiment visade teamet att ALDH4A1 kan bryta ner 4‑HNE direkt, och till skillnad från ett mer känt aldehydenzym, ALDH2, gör det utan att stängas av av just den toxin det försöker undanröja. När forskarna försvagade ALDH4A1 i nervliknande celler blev dessa celler mycket mer sårbara för 4‑HNE: de dog lättare och samlade på sig fler proteinaggregat, vilket tyder på att ALDH4A1 normalt fungerar som en sköld mot denna skadliga molekyl.
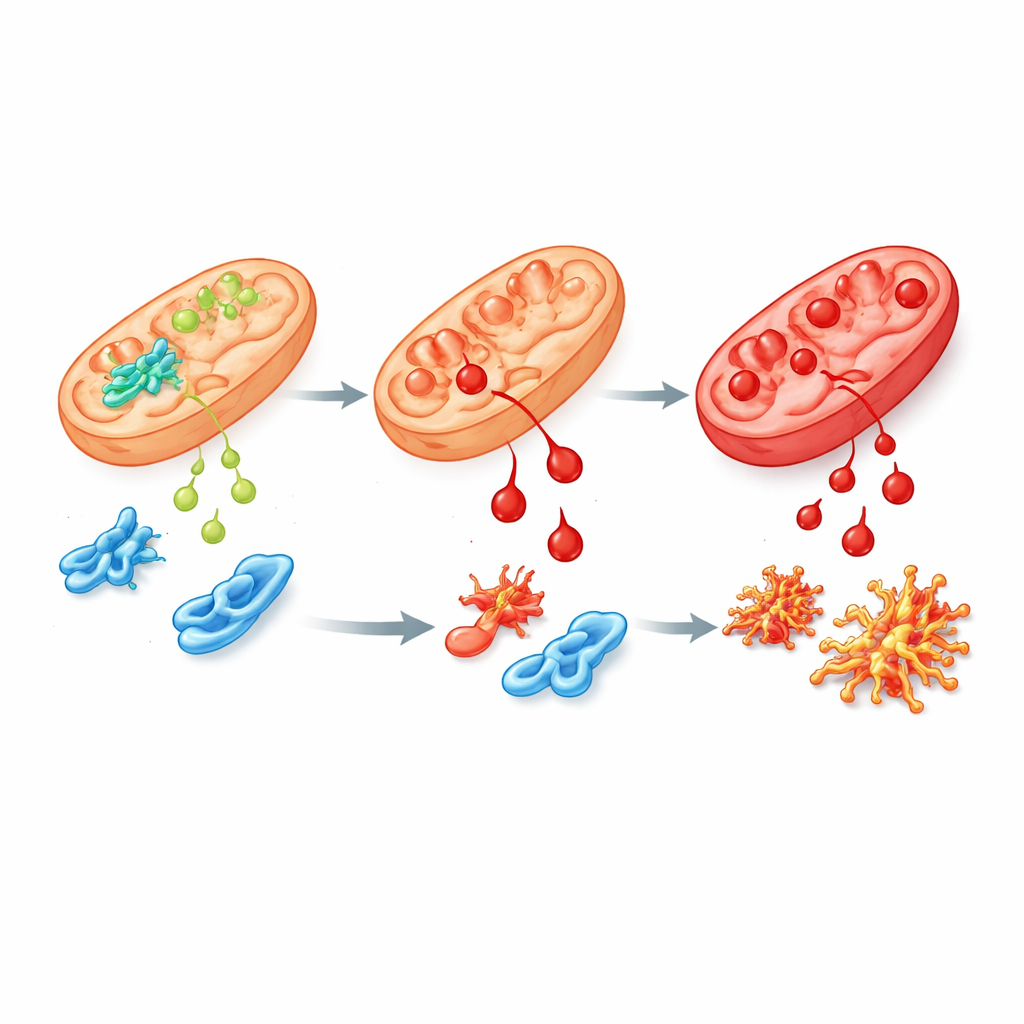
En felaktig variant och dess ringar på vattnet
För att efterlikna sjukdomen hos människor närmare konstruerade teamet humana stamceller och möss med den vanligaste patientmutationen i ALDH4A1, kallad S352L. Denna förändring gör enzymet både mindre aktivt och mycket instabilt, så lite eller inget av det finns kvar i cellerna. I stamceller och i deras laboratoriumodlade nerv‑ och stödjeceller steg prolinhalterna kraftigt, som ses hos patienter. Dessa celler var också mycket mer känsliga för 4‑HNE och ackumulerade kemiska ”ärr” från det snabbare. När forskarna undersökte vilka gener som var på- eller avstängda fann de omfattande störningar i nätverk som styr hjärnans tillväxt, hanterar oxidativ stress och kopplar vitamin B6 till proteiner. Flera proteiner involverade i prolin‑ och polyaminmetabolism, inklusive sperminesynthas, var minskade, vilket antyder en vidgad störning av hjärnans kemi.
Från celler till hela djur
Muss motsvarighet till S352L‑mutationen berättade en liknande historia på organismnivå. Möss med två kopior av den defekta genen hade extremt höga prolinhalter i blod, urin och hjärna, precis som drabbade barn. Många av dessa djur nådde inte vuxen ålder, och kullar från bärare var mindre än väntat, vilket tyder på att mutationen är skadlig redan före födseln. Överlevande möss var mindre, hade lättare hjärnor och visade förändringar i sammansättningen av hjärnceller, med färre astrocyter i förhållande till neuroner. Astrocyter hjälper normalt till att kontrollera den kemiska miljön runt nervceller, inklusive hantering av aminosyror som glutamat och prolin. Resultaten tyder på att när ALDH4A1 är defekt kan astrocyter frisätta extra prolin och misslyckas med att skydda neuroner från aldehydöverbelastning, vilket banar väg för anfall och nedsatt hjärnutveckling.
Varför detta är viktigt bortom en sällsynt sjukdom
Tillsammans omformulerar studien ALDH4A1‑brist som mer än ett enkelt prolinproblem. Förlust av detta enzym förstärker effekten av toxiska aldehyder såsom 4‑HNE, driver proteinaggregation och omprogrammerar genprogram som formar den växande hjärnan. Detta kan förklara varför standardbehandling med vitamin B6 ofta inte räcker och varför patienter fortsätter att ha kognitiva svårigheter. Arbetet väcker också möjligheten att mildare ALDH4A1‑varianter, som är relativt vanliga i befolkningen, tyst kan öka risken för tillstånd kopplade till oxidativ stress och proteinansamling, såsom vissa epilepsier, traumatiska hjärnskador, stroke eller till och med Alzheimers sjukdom. Genom att kartlägga dessa vägar, menar författarna, kan vi börja utforma terapier som stabiliserar eller stärker ALDH4A1, eller som riktar sig mot de efterföljande metaboliska och genuttrycksförändringar det styr.
Citering: Kraemer, B.R., Heo, G., Chen, CH. et al. Increased susceptibility to 4-HNE-induced toxicity and impaired development in a model of ALDH4A1-deficient pediatric epilepsy carrying the S352L variant. Commun Biol 9, 597 (2026). https://doi.org/10.1038/s42003-026-09845-y
Nyckelord: pediatrisk epilepsi, toxiska aldehyder, mitokondriella enzymer, hjärnans utveckling, proteinaggregation