Clear Sky Science · de
Erhöhte Anfälligkeit für 4‑HNE‑induzierte Toxizität und gestörte Entwicklung in einem Modell der ALDH4A1‑defizienten kindlichen Epilepsie mit der S352L‑Variante
Wenn die zelluläre Reinigung versagt
Bei einigen Kindern entwickelt sich schwer behandelbare Epilepsie mit lebenslangen Lernschwierigkeiten aus Gründen, die noch weitgehend ungeklärt sind. Diese Studie untersucht eine solche Ursache: seltene Veränderungen in einem einzelnen Enzym innerhalb der Kraftwerke unserer Zellen, den Mitochondrien. Indem die Forscherinnen und Forscher erforschen, wie dieses Enzym normalerweise toxische Nebenprodukte entfernt und was geschieht, wenn es ausfällt, liefern sie neue Hinweise darauf, warum Anfälle entstehen, warum das Gehirn sich möglicherweise nicht richtig entwickelt und wie wir bessere Behandlungen entwerfen könnten.
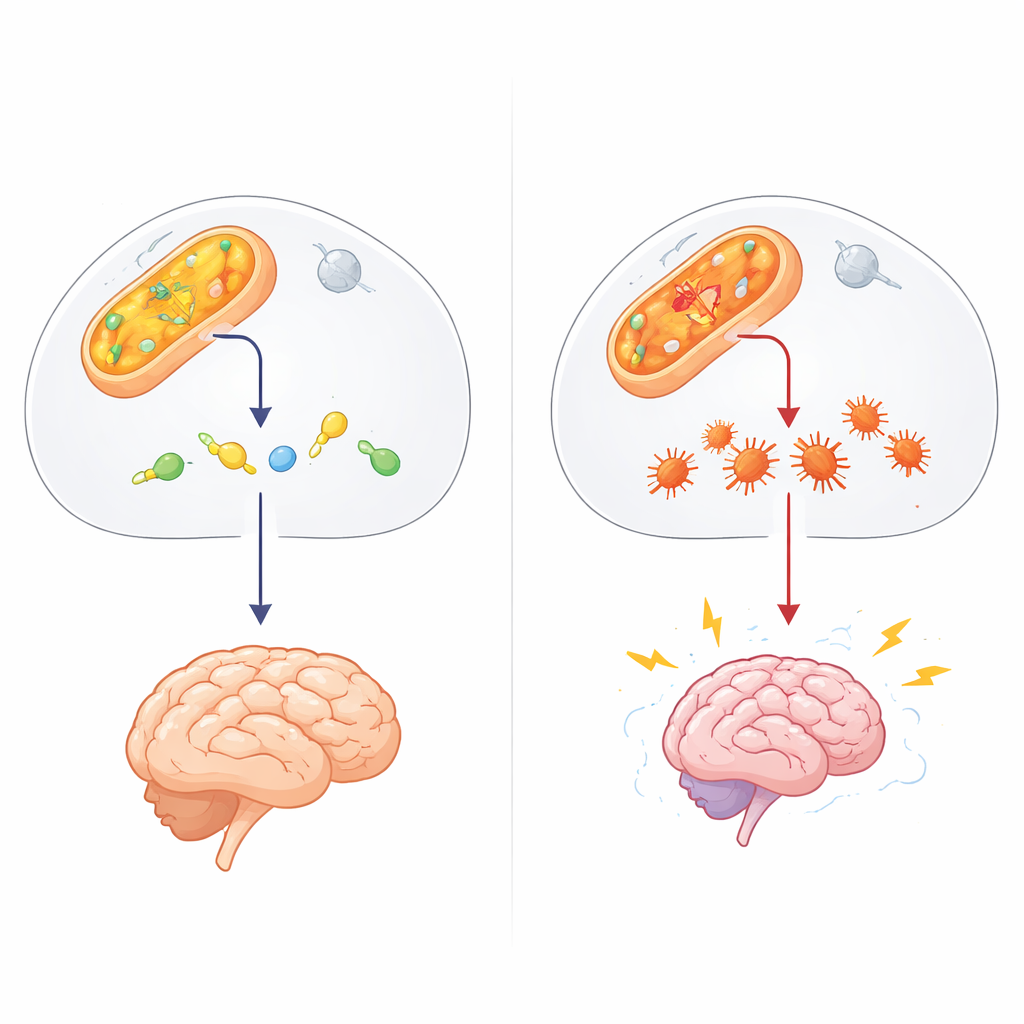
Ein verborgener Wächter der Zellchemie
Die Arbeit konzentriert sich auf ALDH4A1, ein Mitglied einer großen Enzymfamilie, die Aldehyde entgiftet — hochreaktive chemische Überreste, die bei normalem Stoffwechsel, durch Umwelteinflüsse und manche Medikamente entstehen. ALDH4A1 baut gewöhnlich eine Verbindung ab, die mit der Aminosäure Prolin verknüpft ist, und trägt so dazu bei, Vitamin B6 aktiv zu halten. Fehlt ALDH4A1 oder ist es fehlerhaft, entwickeln Betroffene die seltene Kindkrankheit Hyperprolinämie Typ II, gekennzeichnet durch sehr hohe Prolinspiegel, Anfälle und Entwicklungsverzögerung. Bislang wurden die meisten Erklärungen auf überschüssiges Prolin und einen Mangel an Vitamin B6 zurückgeführt. Diese Ideen konnten jedoch Patienten, die auf Vitaminergänzung nicht ansprechen, oder die rätselhaften Unterschiede zwischen verwandten Erkrankungen nicht vollständig erklären.
Ein toxisches Molekül im Rampenlicht
Die Autorinnen und Autoren vermuteten einen weiteren Beteiligten: 4‑Hydroxynonenal (4‑HNE), ein klebriger Aldehyd, der entsteht, wenn Fette in Zellmembranen durch oxidativen Stress, etwa während Anfällen, beschädigt werden. 4‑HNE kann sich an Proteine und DNA binden und deren Fehlfaltung und Verklebung verursachen. In Reagenzglas‑Experimenten zeigten die Forschenden, dass ALDH4A1 4‑HNE direkt abbauen kann, und im Gegensatz zu dem bekannteren Aldehyd‑Enzym ALDH2 geschieht dies, ohne dass ALDH4A1 durch das Toxin selbst blockiert wird. Schwächten die Forschenden ALDH4A1 in nervenähnlichen Zellen, wurden diese Zellen deutlich empfindlicher gegenüber 4‑HNE: sie starben leichter und häuften mehr Proteinaggregate an, was darauf hindeutet, dass ALDH4A1 normalerweise als Schutzschild gegen dieses schädigende Molekül wirkt.
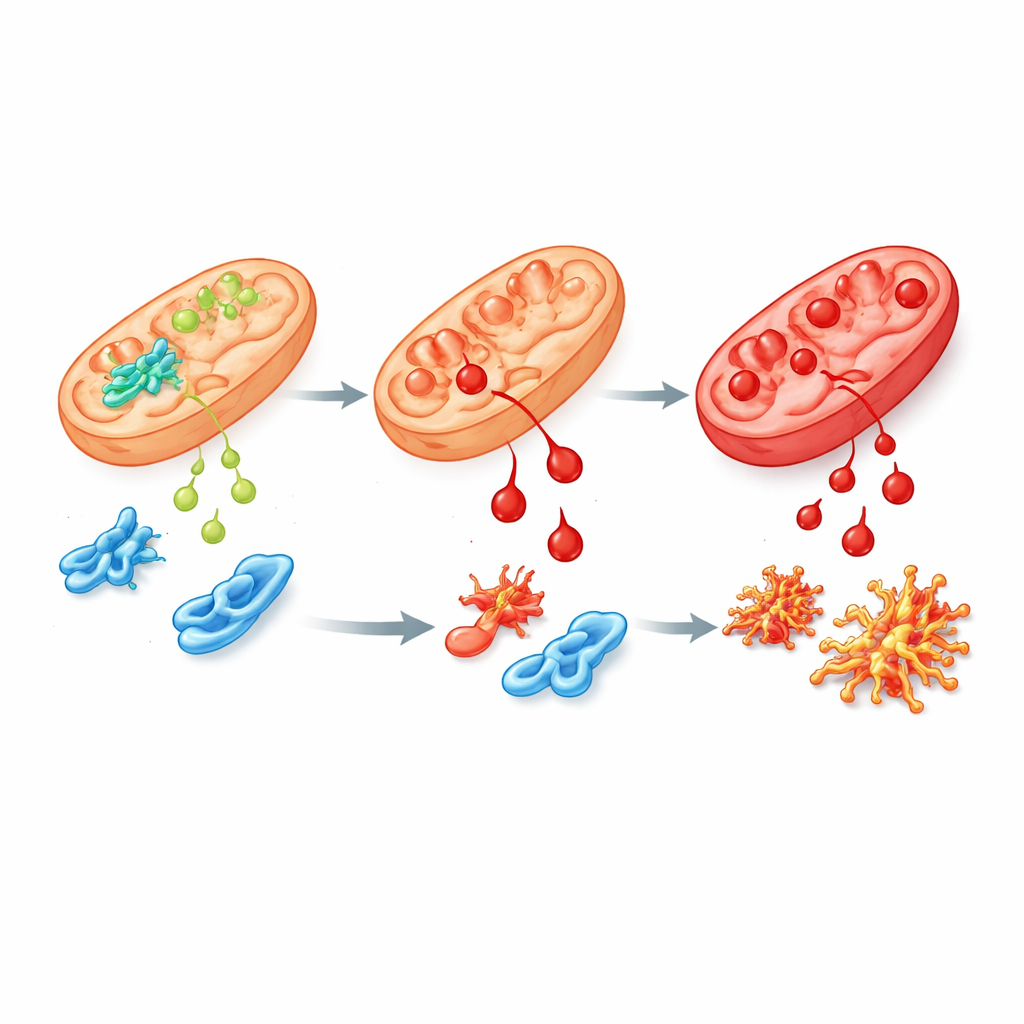
Eine fehlerhafte Variante und ihre Kaskadenwirkung
Um die menschliche Erkrankung näher zu modellieren, erzeugte das Team humane Stammzellen und Mäuse mit der häufigsten Patientenmutation in ALDH4A1, genannt S352L. Diese Veränderung macht das Enzym sowohl weniger aktiv als auch sehr instabil, sodass nur wenig oder gar kein Enzym in Zellen verbleibt. In Stammzellen sowie in ihren im Labor gezüchteten Nerven‑ und Stützzellen stiegen die Prolinspiegel stark an, wie bei betroffenen Patienten. Diese Zellen waren zudem deutlich empfindlicher gegenüber 4‑HNE und häuften schneller chemische »Narben« durch dieses Molekül an. Bei der Untersuchung der Genaktivität zeigten sich weitreichende Störungen in Netzwerken, die das Gehirnwachstum steuern, oxidativen Stress bewältigen und Vitamin‑B6‑Verknüpfungen mit Proteinen regeln. Mehrere Proteine, die an Prolin‑ und Polyamin‑Stoffwechsel beteiligt sind, darunter die Sperminsynthase, waren reduziert, was auf eine umfassendere Beeinträchtigung der Hirnchemie hindeutet.
Von Zellen zu ganzen Tieren
Die Mäuse mit der S352L‑Mutation erzählten eine ähnliche Geschichte auf Körperebene. Mäuse mit zwei Kopien des fehlerhaften Gens wiesen extrem hohe Prolinspiegel im Blut, Urin und Gehirn auf, wie betroffene Kinder. Viele dieser Tiere überlebten nicht bis ins Erwachsenenalter, und die Wurfgrößen tragender Elternpaare waren kleiner als erwartet, was darauf hindeutet, dass die Mutation bereits vor der Geburt schädlich ist. Überlebende Mäuse waren kleiner, hatten leichtere Gehirne und zeigten Verschiebungen in der Zusammensetzung der Gehirnzellen, mit weniger Astrozyten im Verhältnis zu Neuronen. Astrozyten regulieren normalerweise das chemische Umfeld der Nervenzellen, einschließlich des Umgangs mit Aminosäuren wie Glutamat und Prolin. Die Befunde legen nahe, dass bei defektem ALDH4A1 Astrozyten möglicherweise zusätzliches Prolin freisetzen und die Neuronen nicht ausreichend vor Aldehydüberladung schützen, was den Nährboden für Anfälle und mangelhafte Gehirnentwicklung bereitet.
Warum das über eine seltene Krankheit hinaus relevant ist
In der Summe stellt die Studie ALDH4A1‑Mangel als mehr dar als ein reines Prolinproblem. Der Verlust dieses Enzyms verstärkt die Wirkung toxischer Aldehyde wie 4‑HNE, treibt die Proteinaggregation voran und verändert Genprogramme, die das reifende Gehirn formen. Das könnte erklären, warum die übliche Vitamin‑B6‑Therapie oft nicht ausreicht und weshalb Betroffene weiter kognitive Probleme haben. Die Arbeit weckt außerdem die Möglichkeit, dass mildere ALDH4A1‑Varianten, die in der Bevölkerung relativ häufig vorkommen, still das Risiko für Erkrankungen erhöhen könnten, die mit oxidativem Stress und Proteinablagerungen zusammenhängen — etwa bestimmte Epilepsien, Schädel‑Hirn‑Trauma, Schlaganfall oder sogar Alzheimer. Indem diese Signalwege kartiert werden, argumentieren die Autorinnen und Autoren, lasse sich anfangen, Therapien zu entwickeln, die ALDH4A1 stabilisieren oder stärken oder die auf die nachgelagerten Stoffwechsel‑ und Genexpressionsänderungen abzielen.
Zitation: Kraemer, B.R., Heo, G., Chen, CH. et al. Increased susceptibility to 4-HNE-induced toxicity and impaired development in a model of ALDH4A1-deficient pediatric epilepsy carrying the S352L variant. Commun Biol 9, 597 (2026). https://doi.org/10.1038/s42003-026-09845-y
Schlüsselwörter: pädiatrische Epilepsie, toxische Aldehyde, mitochondriale Enzyme, Gehirnentwicklung, Proteinaggregation