Clear Sky Science · pl
Zwiększona podatność na toksyczne działanie 4-HNE i upośledzony rozwój w modelu pediatrycznej padaczki związanej z niedoborem ALDH4A1 niosącym wariant S352L
Gdy komórkowy zespół sprzątający zawodzi
U niektórych dzieci występuje oporna na leczenie padaczka i trwałe trudności w uczeniu się z przyczyn wciąż nie w pełni poznanych. W tym badaniu analizowano jedną z takich przyczyn: rzadkie zmiany w pojedynczym enzymie wewnątrz „elektrowni” komórek — mitochondriów. Poprzez badanie, jak ten enzym zwykle usuwa toksyczne produkty uboczne i co się dzieje, gdy zawodzi, autorzy ujawniają nowe wskazówki dotyczące tego, dlaczego pojawiają się napady, dlaczego mózg może rozwijać się nieprawidłowo i jak można projektować skuteczniejsze terapie.
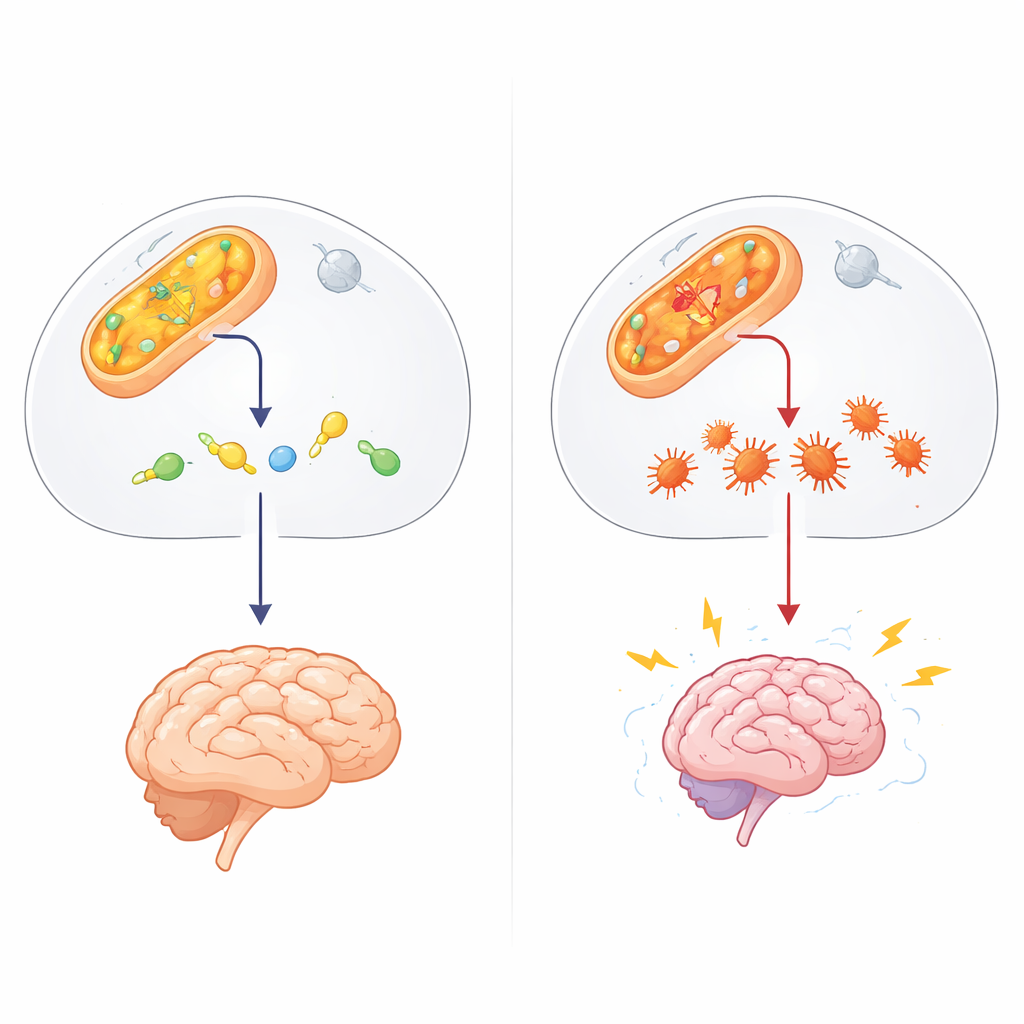
Ukryty strażnik w chemii komórkowej
Praca koncentruje się na ALDH4A1, należącym do dużej rodziny enzymów detoksykujących aldehydy — wysoce reaktywne cząsteczki powstające podczas normalnego metabolizmu, ekspozycji środowiskowej i w efekcie niektórych leków. ALDH4A1 zwykle rozkłada związek związany z aminokwasem prolina, pomagając utrzymać aktywność witaminy B6. Gdy ALDH4A1 brakują lub jest nieprawidłowy, u chorych rozwija się hiperprolinemia typu II, rzadka choroba dziecięca charakteryzująca się bardzo wysokim stężeniem proliny, napadami i opóźnieniem rozwoju. Dotychczas większość wyjaśnień obwiniała nadmiar proliny i niedobór witaminy B6. Jednak te hipotezy nie tłumaczyły w pełni pacjentów, którzy nie poprawiają się po suplementach witaminy, ani zagadkowych różnic między powiązanymi zaburzeniami.
Toksyczna cząsteczka na świeczniku
Autorzy podejrzewali udział innego czynnika: 4-hydroksynonenalu (4-HNE), lepkiego aldehydu powstającego, gdy tłuszcze błon komórkowych są uszkadzane przez stres oksydacyjny, na przykład podczas napadów. 4-HNE może przyłączać się do białek i DNA, powodując ich nieprawidłowe fałdowanie i tworzenie grudek. W eksperymentach in vitro zespół wykazał, że ALDH4A1 może bezpośrednio rozkładać 4-HNE, i w odróżnieniu od lepiej znanego enzymu aldehydowego ALDH2 robi to bez ulegania zahamowaniu przez samą toksynę, którą powinien usuwać. Gdy badacze osłabili ALDH4A1 w komórkach o charakterze nerwowym, komórki te stały się znacznie bardziej wrażliwe na 4-HNE: częściej umierały i gromadziły więcej agregatów białkowych, co sugeruje, że ALDH4A1 normalnie działa jak tarcza przeciwko tej szkodliwej cząsteczce.
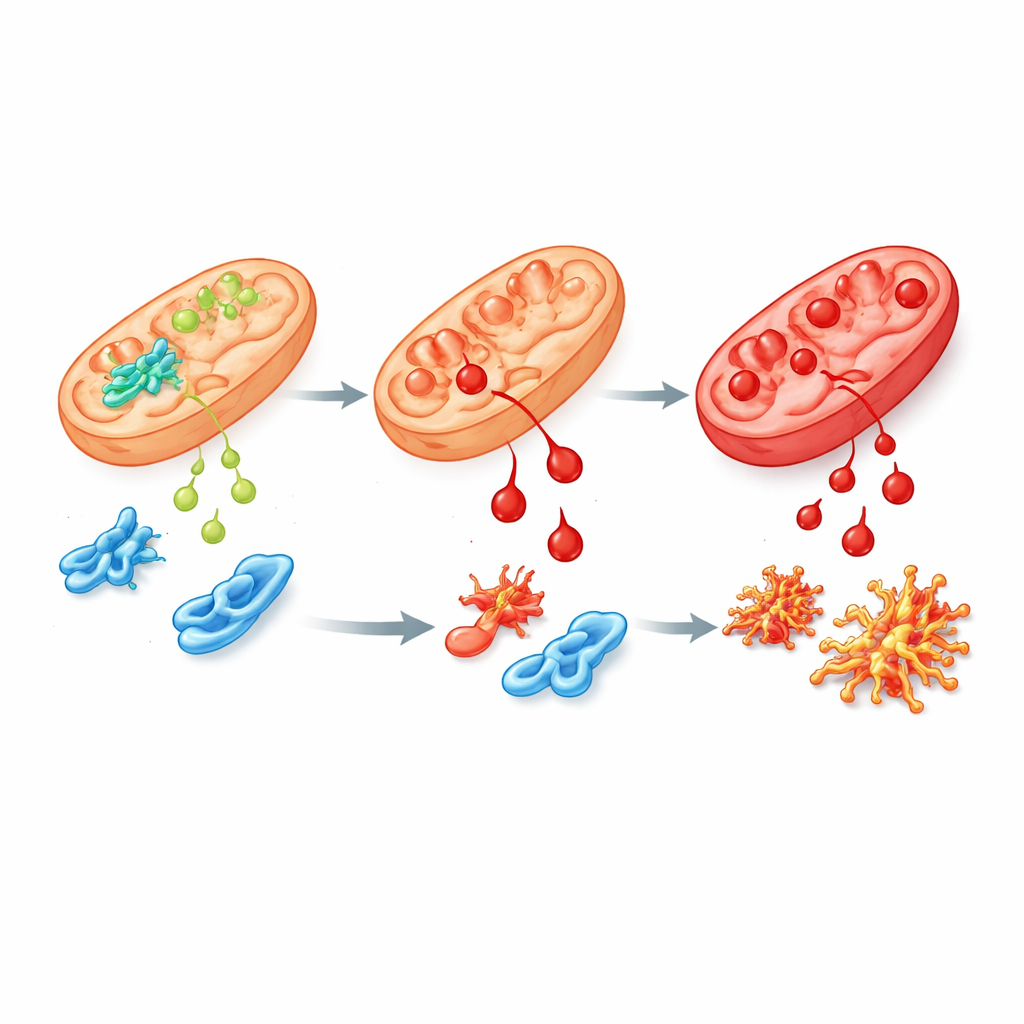
Uszkodzony wariant i jego skutki
Aby lepiej odwzorować ludzką chorobę, zespół wprowadził do ludzkich komórek macierzystych i myszy najczęściej występującą u pacjentów mutację w ALDH4A1, nazwaną S352L. Zmiana ta sprawia, że enzym jest zarówno mniej aktywny, jak i bardzo niestabilny, więc w komórkach pozostaje go bardzo mało lub wcale. W komórkach macierzystych oraz w wyhodowanych w laboratorium komórkach nerwowych i podporowych stężenie proliny gwałtownie wzrosło, jak obserwuje się u pacjentów. Te komórki były też znacznie bardziej wrażliwe na 4-HNE i szybciej gromadziły chemiczne „blizny” po nim. Analiza ekspresji genów wykazała szerokie zaburzenia w sieciach kierujących wzrostem mózgu, radzeniem sobie ze stresem oksydacyjnym oraz powiązaniami witaminy B6 z białkami. Kilka białek zaangażowanych w metabolizm proliny i poliamin, w tym syntaza spermyny, było zmniejszonych, sugerując szersze zaburzenia chemii mózgu.
Od komórek do całego zwierzęcia
Mysia wersja mutacji S352L opowiadała podobną historię na poziomie całego organizmu. Myszy z dwiema kopiami wadliwego genu miały niezwykle wysokie stężenia proliny we krwi, moczu i mózgu, podobnie jak dotknięte chorobą dzieci. Wiele z tych zwierząt nie przeżywało do dorosłości, a mioty od rodziców-nosicieli były mniejsze niż oczekiwano, co sugeruje, że mutacja szkodzi już przed narodzinami. Przeżywające myszy były mniejsze, miały lżejsze mózgi i wykazywały zmiany w składzie komórek mózgowych — mniej astrocytów w stosunku do neuronów. Astrocyty zwykle pomagają kontrolować środowisko chemiczne wokół komórek nerwowych, w tym gospodarowanie aminokwasami takimi jak glutaminian i prolina. Wyniki sugerują, że gdy ALDH4A1 jest nieprawidłowy, astrocyty mogą uwalniać nadmiar proliny i nie chronić neuronów przed przeciążeniem aldehydami, tworząc podłoże do napadów i zaburzeń rozwoju mózgu.
Dlaczego to ma znaczenie poza rzadką chorobą
Podsumowując, badanie ukazuje niedobór ALDH4A1 jako coś więcej niż prosty problem z proliną. Utrata tego enzymu wzmacnia działanie toksycznych aldehydów takich jak 4-HNE, napędza agregację białek i przestawia programy genowe kształtujące rozwijający się mózg. To może wyjaśniać, dlaczego standardowa terapia witaminą B6 często zawodzi i dlaczego pacjenci nadal mają trudności poznawcze. Praca sugeruje także możliwość, że łagodniejsze warianty ALDH4A1, stosunkowo częste w populacji, mogą po cichu zwiększać ryzyko stanów związanych ze stresem oksydacyjnym i kumulacją białek, takich jak niektóre postacie padaczki, urazy mózgu, udar, a nawet choroba Alzheimera. Mapując te ścieżki, autorzy twierdzą, że można zacząć projektować terapie stabilizujące lub wspomagające ALDH4A1 albo celujące w wtórne zmiany metaboliczne i ekspresji genów, które ten enzym kontroluje.
Cytowanie: Kraemer, B.R., Heo, G., Chen, CH. et al. Increased susceptibility to 4-HNE-induced toxicity and impaired development in a model of ALDH4A1-deficient pediatric epilepsy carrying the S352L variant. Commun Biol 9, 597 (2026). https://doi.org/10.1038/s42003-026-09845-y
Słowa kluczowe: padaczka dziecięca, toksyczne aldehydy, enzymy mitochondrialne, rozwój mózgu, agregacja białek