Clear Sky Science · it
Aumentata suscettibilità alla tossicità indotta da 4-HNE e sviluppo compromesso in un modello di epilessia pediatrica da deficit di ALDH4A1 con la variante S352L
Quando la squadra di pulizia della cellula fallisce
Alcuni bambini sviluppano epilessia difficile da curare e problemi di apprendimento che durano tutta la vita per ragioni ancora in gran parte sconosciute. Questo studio analizza una di queste cause: variazioni rare in un singolo enzima all'interno delle centrali energetiche delle nostre cellule, i mitocondri. Esaminando come questo enzima normalmente contribuisce a eliminare prodotti di scarto tossici e cosa succede quando non funziona, i ricercatori scoprono nuovi indizi sul perché iniziano le crisi, perché il cervello può non svilupparsi correttamente e come si potrebbero progettare terapie migliori.
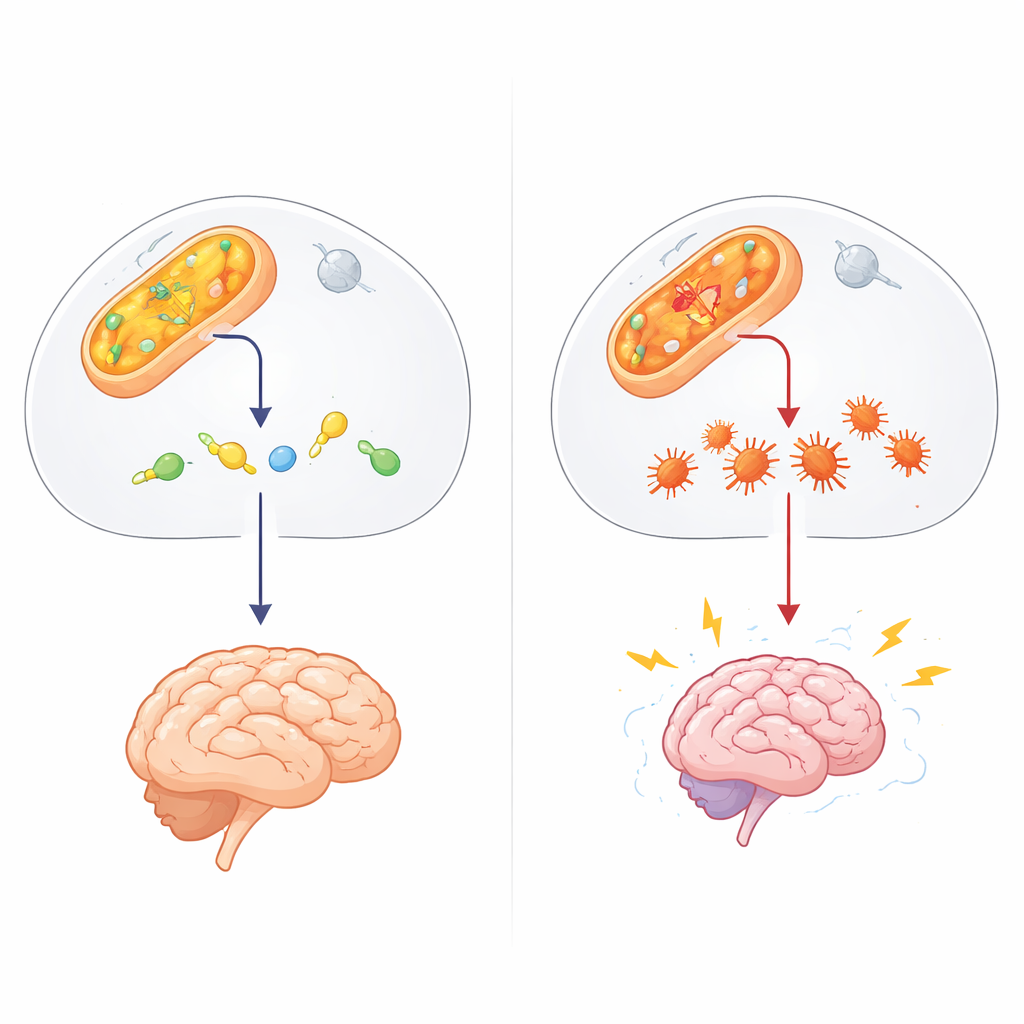
Un guardiano nascosto nella chimica cellulare
Il lavoro si concentra su ALDH4A1, parte di una vasta famiglia di enzimi che detossificano le aldeidi—scarti chimici altamente reattivi prodotti dal metabolismo normale, da esposizioni ambientali e da alcuni farmaci. ALDH4A1 normalmente scompone un composto legato all'amminoacido prolina, contribuendo a mantenere attivo il vitamina B6. Quando ALDH4A1 manca o è difettoso, le persone sviluppano l'ipertroprolinemia di tipo II, una malattia infantile rara caratterizzata da livelli molto elevati di prolina, crisi epilettiche e ritardo nello sviluppo. Finora, la maggior parte delle spiegazioni ha attribuito il quadro all'eccesso di prolina e alla carenza di vitamina B6. Tuttavia, queste idee non riuscivano a spiegare completamente i pazienti che non migliorano con gli integratori vitaminici o le differenze enigmatiche tra disturbi correlati.
Una molecola tossica sotto i riflettori
Gli autori hanno sospettato l'intervento di un altro protagonista: il 4-idrossinonennale (4-HNE), un'aldeide appiccicosa che si forma quando i grassi nelle membrane cellulari vengono danneggiati dallo stress ossidativo, ad esempio durante le crisi. Il 4-HNE può legarsi a proteine e DNA, facendoli ripiegare in modo errato e aggregare. In esperimenti in provetta, il gruppo ha dimostrato che ALDH4A1 può degradare direttamente il 4-HNE e, a differenza di un enzima delle aldeidi più noto, ALDH2, lo fa senza venire bloccato dal tossico che cerca di rimuovere. Quando i ricercatori hanno indebolito ALDH4A1 in cellule di tipo nervoso, quelle cellule sono diventate molto più vulnerabili al 4-HNE: morivano più facilmente e accumulavano più aggregati proteici, suggerendo che ALDH4A1 agisca normalmente come scudo contro questa molecola dannosa.
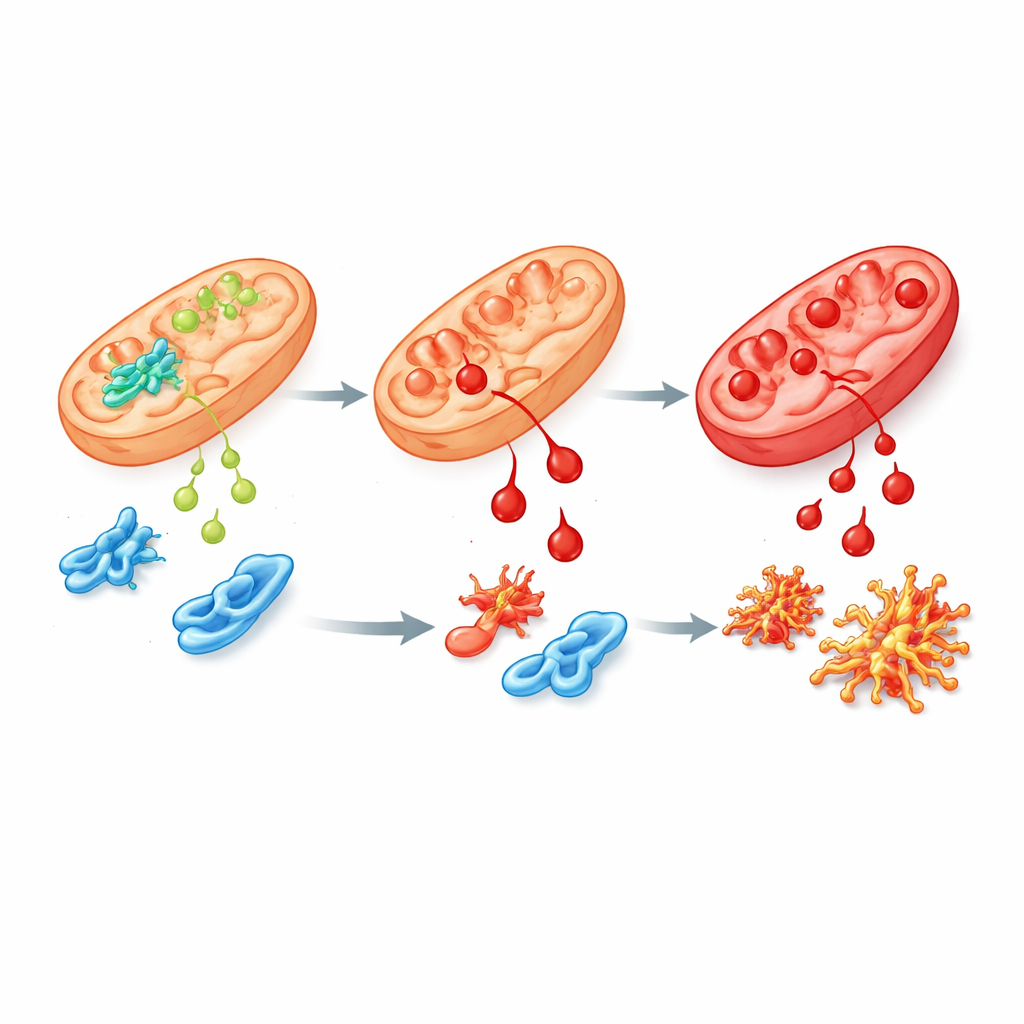
Una variante difettosa e i suoi effetti a catena
Per imitare più da vicino la malattia umana, il team ha ingegnerizzato cellule staminali umane e topi portatori della mutazione più comune dei pazienti in ALDH4A1, chiamata S352L. Questo cambiamento rende l'enzima sia meno attivo sia altamente instabile, così che ne rimane poco o nulla nelle cellule. In cellule staminali e nelle cellule nervose e di supporto coltivate in laboratorio, i livelli di prolina sono aumentati marcatamente, come osservato nei pazienti. Queste cellule erano inoltre molto più sensibili al 4-HNE e accumulavano più rapidamente "cicatrici" chimiche dovute ad esso. Quando i ricercatori hanno esaminato quali geni risultavano attivati o silenziati, hanno trovato ampie perturbazioni nelle reti che guidano la crescita cerebrale, gestiscono lo stress ossidativo e collegano la vitamina B6 alle proteine. Diverse proteine coinvolte nel metabolismo della prolina e delle poliammine, inclusa la spermine sintasi, risultavano ridotte, suggerendo una più ampia alterazione della chimica cerebrale.
Dalle cellule agli animali interi
La versione murina della mutazione S352L ha raccontato una storia simile a livello dell'intero organismo. I topi con due copie del gene difettoso presentavano livelli estremamente alti di prolina nel sangue, nelle urine e nel cervello, proprio come i bambini colpiti. Molti di questi animali non raggiungevano l'età adulta, e le cucciolate provenienti da genitori portatori erano più piccole del previsto, suggerendo che la mutazione è dannosa già prima della nascita. I topi sopravvissuti erano più piccoli, avevano cervelli più leggeri e mostravano cambiamenti nella composizione cellulare cerebrale, con meno astrociti rispetto ai neuroni. Normalmente gli astrociti aiutano a controllare l'ambiente chimico attorno alle cellule nervose, inclusa la gestione di amminoacidi come glutammato e prolina. I risultati suggeriscono che quando ALDH4A1 è difettoso, gli astrociti possono rilasciare prolina in eccesso e non riuscire a proteggere i neuroni dal sovraccarico di aldeidi, creando le condizioni per crisi epilettiche e scarso sviluppo cerebrale.
Perché questo conta oltre una malattia rara
Mettendo insieme i risultati, lo studio reinterpreta il deficit di ALDH4A1 come qualcosa di più di un semplice problema di prolina. La perdita di questo enzima amplifica l'impatto di aldeidi tossiche come il 4-HNE, favorisce l'aggregazione proteica e riorganizza programmi genici che modellano il cervello in crescita. Questo potrebbe spiegare perché la terapia standard con vitamina B6 spesso non è sufficiente e perché i pazienti continuano ad avere difficoltà cognitive. Il lavoro solleva anche la possibilità che varianti più lievi di ALDH4A1, relativamente comuni nella popolazione, possano aumentare silenziosamente il rischio per condizioni legate allo stress ossidativo e all'accumulo proteico, come alcune epilessie, traumi cranici, ictus o persino il morbo di Alzheimer. Mappando queste vie, gli autori sostengono che si possono iniziare a progettare terapie che stabilizzino o potenzino ALDH4A1, o che prendano di mira i cambiamenti metabolici e di espressione genica a valle che essa controlla.
Citazione: Kraemer, B.R., Heo, G., Chen, CH. et al. Increased susceptibility to 4-HNE-induced toxicity and impaired development in a model of ALDH4A1-deficient pediatric epilepsy carrying the S352L variant. Commun Biol 9, 597 (2026). https://doi.org/10.1038/s42003-026-09845-y
Parole chiave: epilessia pediatrica, aldeidi tossiche, enzimi mitocondriali, sviluppo cerebrale, aggregazione proteica