Clear Sky Science · pt
Maior suscetibilidade à toxicidade induzida por 4-HNE e desenvolvimento prejudicado em um modelo de epilepsia pediátrica com deficiência de ALDH4A1 portador da variante S352L
Quando a equipe de limpeza da célula falha
Algumas crianças desenvolvem epilepsia de difícil tratamento e dificuldades de aprendizagem ao longo da vida por razões que ainda são misteriosas. Este estudo investiga uma dessas causas: alterações raras em uma única enzima nas usinas de energia das nossas células, as mitocôndrias. Ao explorar como essa enzima normalmente ajuda a eliminar subprodutos tóxicos e o que ocorre quando ela falha, os pesquisadores descobrem novas pistas sobre por que as convulsões começam, por que o cérebro pode não se desenvolver adequadamente e como poderíamos projetar tratamentos melhores.
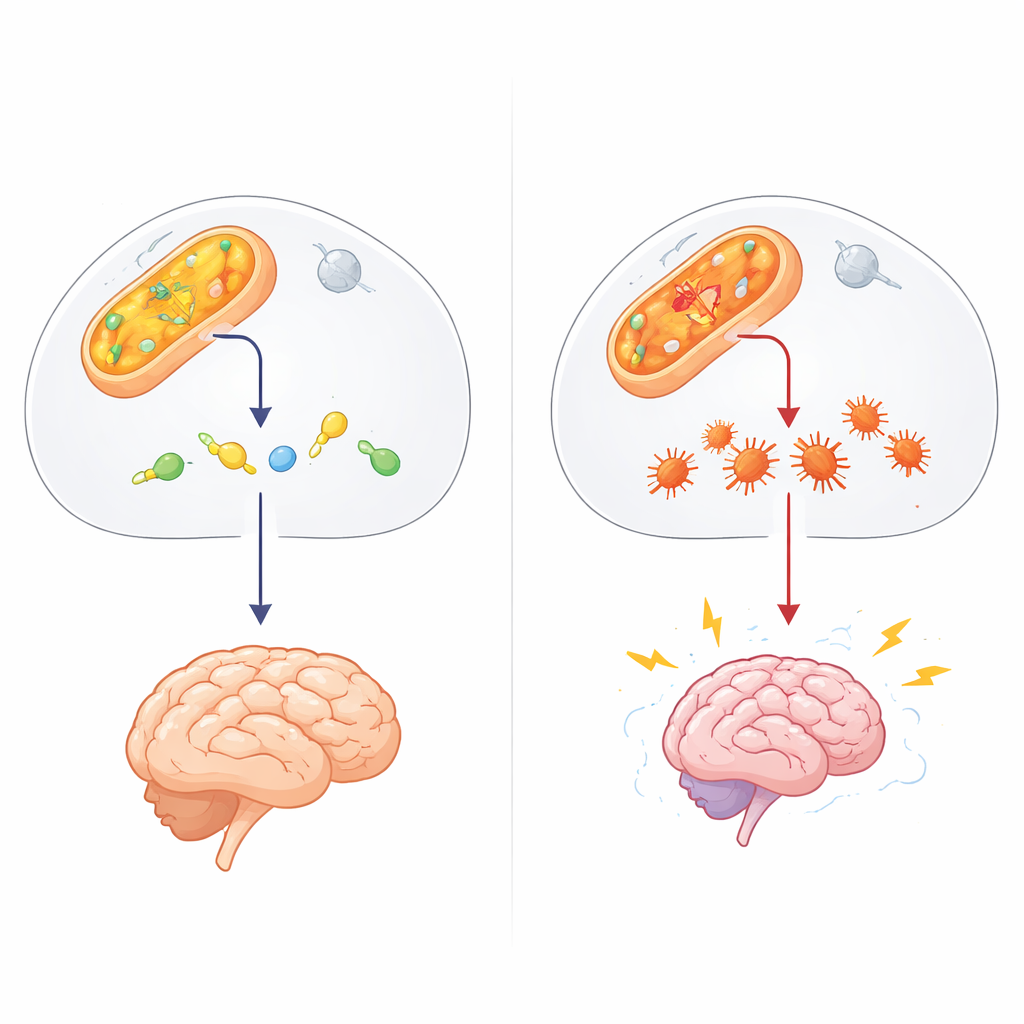
Um guardião oculto na química celular
O trabalho centra-se na ALDH4A1, parte de uma grande família de enzimas que desintoxicam aldeídos — resíduos químicos altamente reativos produzidos pelo metabolismo normal, exposições ambientais e alguns medicamentos. A ALDH4A1 normalmente degrada um composto relacionado ao aminoácido prolina, ajudando a manter a vitamina B6 ativa. Quando a ALDH4A1 está ausente ou defeituosa, as pessoas desenvolvem hiperprolinemia tipo II, uma doença infantil rara marcada por níveis muito altos de prolina, convulsões e atraso do desenvolvimento. Até agora, a maioria das explicações atribuía o quadro ao excesso de prolina e à falta de vitamina B6. No entanto, essas ideias não explicavam totalmente pacientes que não melhoram com suplementos vitamínicos ou as diferenças intrigantes entre distúrbios relacionados.
Uma molécula tóxica em foco
Os autores suspeitaram de outro ator: o 4-hidroxinonenal (4-HNE), um aldeído pegajoso formado quando gorduras nas membranas celulares são danificadas pelo estresse oxidativo, como durante convulsões. O 4-HNE pode ligar-se a proteínas e DNA, fazendo com que se dobrem incorretamente e se aglomerem. Em experimentos de tubo de ensaio, a equipe mostrou que a ALDH4A1 pode degradar diretamente o 4-HNE e, ao contrário de uma enzima de aldeído mais conhecida, a ALDH2, faz isso sem ser inativada pelo próprio tóxico que tenta remover. Quando os pesquisadores enfraqueceram a ALDH4A1 em células do tipo nervoso, essas células tornaram-se muito mais vulneráveis ao 4-HNE: morreram com mais facilidade e acumularam mais agregados de proteínas, sugerindo que a ALDH4A1 normalmente atua como um escudo contra essa molécula danosa.
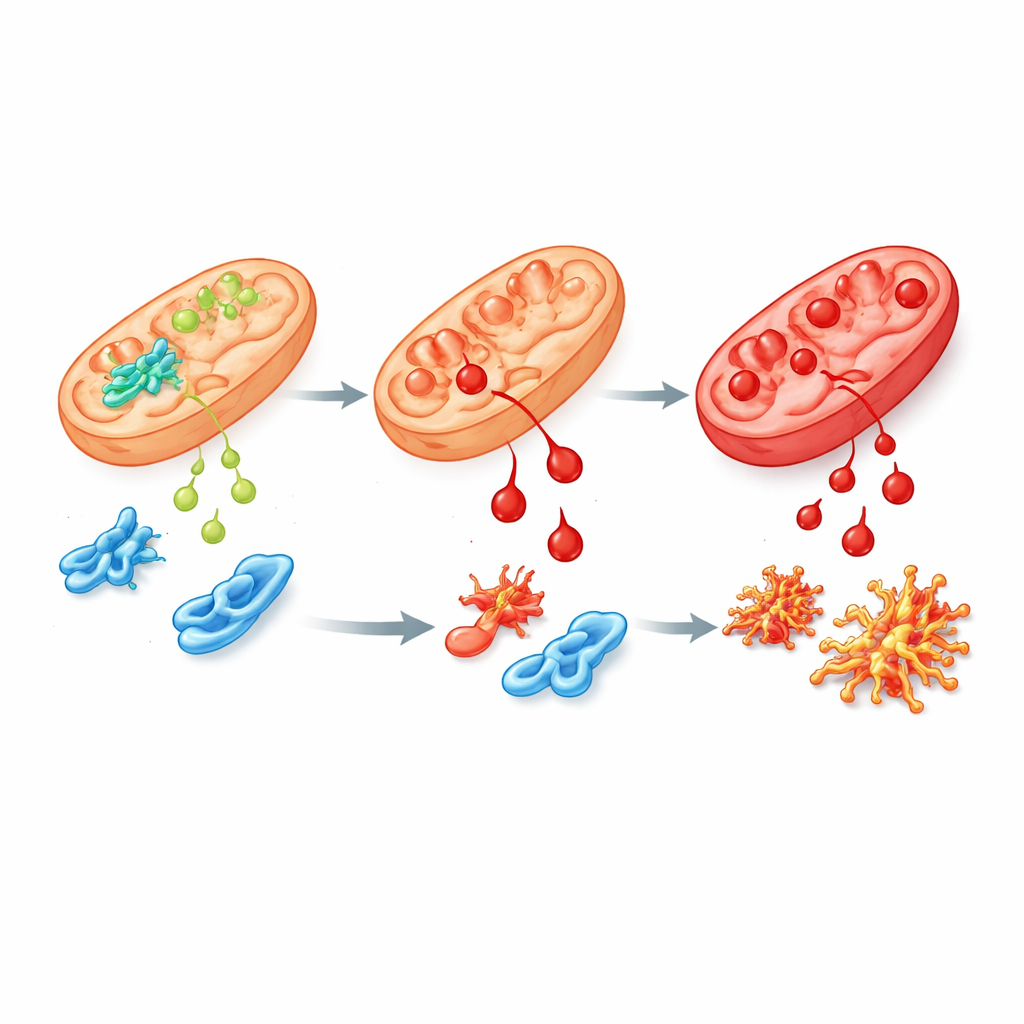
Uma variante defeituosa e seus efeitos em cascata
Para imitar mais de perto a doença humana, a equipe modificou células-tronco humanas e camundongos para portar a mutação mais comum de pacientes na ALDH4A1, chamada S352L. Essa alteração torna a enzima menos ativa e altamente instável, de modo que pouco ou nenhum resquício dela permanece nas células. Em células-tronco e em suas células nervosas e de suporte cultivadas em laboratório, os níveis de prolina aumentaram acentuadamente, como observado em pacientes. Essas células também foram muito mais sensíveis ao 4-HNE e acumularam “cicatrizes” químicas dele mais rapidamente. Ao examinar quais genes estavam ligados ou desligados, os pesquisadores encontraram perturbações amplas em redes que orientam o crescimento cerebral, lidam com o estresse oxidativo e conectam a vitamina B6 às proteínas. Várias proteínas envolvidas no metabolismo da prolina e das poliaminas, incluindo a espermina sintase, estavam reduzidas, sugerindo uma interrupção mais ampla da química cerebral.
Das células aos organismos inteiros
A versão murina da mutação S352L contou uma história semelhante no nível do organismo. Camundongos com duas cópias do gene defeituoso apresentaram níveis extremamente altos de prolina no sangue, na urina e no cérebro, exatamente como crianças afetadas. Muitos desses animais não sobreviveram até a vida adulta, e ninhadas de pais portadores foram menores do que o esperado, sugerindo que a mutação é prejudicial mesmo antes do nascimento. Os camundongos sobreviventes eram menores, apresentavam cérebros mais leves e mostraram alterações na composição celular do cérebro, com menos astrócitos em relação aos neurônios. Astrócitos normalmente ajudam a controlar o ambiente químico ao redor das células nervosas, incluindo o manejo de aminoácidos como glutamato e prolina. Os achados sugerem que, quando a ALDH4A1 é defeituosa, os astrócitos podem liberar prolina extra e falhar em proteger os neurônios do excesso de aldeídos, preparando o terreno para convulsões e desenvolvimento cerebral pobre.
Por que isso importa além de uma doença rara
Em conjunto, o estudo reconstrói a deficiência de ALDH4A1 como algo além de um simples problema de prolina. A perda dessa enzima amplifica o impacto de aldeídos tóxicos como o 4-HNE, promove agregação de proteínas e reprograma circuitos gênicos que moldam o cérebro em crescimento. Isso pode explicar por que a terapia padrão com vitamina B6 muitas vezes é insuficiente e por que os pacientes continuam a ter dificuldades cognitivas. O trabalho também levanta a possibilidade de que variantes mais brandas de ALDH4A1, relativamente comuns na população, possam aumentar silenciosamente o risco de condições ligadas ao estresse oxidativo e ao acúmulo de proteínas, como certas epilepsias, traumatismo cranioencefálico, AVC ou até mesmo doença de Alzheimer. Ao mapear essas vias, argumentam os autores, podemos começar a projetar terapias que estabilizem ou aumentem a ALDH4A1, ou que atuem nas alterações metabólicas e de expressão gênica que ela controla.
Citação: Kraemer, B.R., Heo, G., Chen, CH. et al. Increased susceptibility to 4-HNE-induced toxicity and impaired development in a model of ALDH4A1-deficient pediatric epilepsy carrying the S352L variant. Commun Biol 9, 597 (2026). https://doi.org/10.1038/s42003-026-09845-y
Palavras-chave: epilepsia pediátrica, aldeídos tóxicos, enzimas mitocondriais, desenvolvimento cerebral, agregação de proteínas