Clear Sky Science · fr
Susceptibilité accrue à la toxicité induite par le 4‑HNE et développement altéré dans un modèle d’épilepsie pédiatrique liée à une déficience en ALDH4A1 portant la variante S352L
Quand l’équipe de nettoyage de la cellule faillit
Certaines enfants développent une épilepsie difficile à traiter et des troubles durables de l’apprentissage pour des raisons encore mal comprises. Cette étude examine l’une de ces causes : des altérations rares d’une enzyme unique située dans les centrales énergétiques de nos cellules, les mitochondries. En explorant comment cette enzyme aide normalement à éliminer des sous‑produits toxiques et ce qui arrive lorsqu’elle dysfonctionne, les chercheurs mettent en lumière de nouveaux indices sur l’origine des crises, sur les raisons d’un développement cérébral perturbé et sur les pistes pour concevoir de meilleurs traitements.
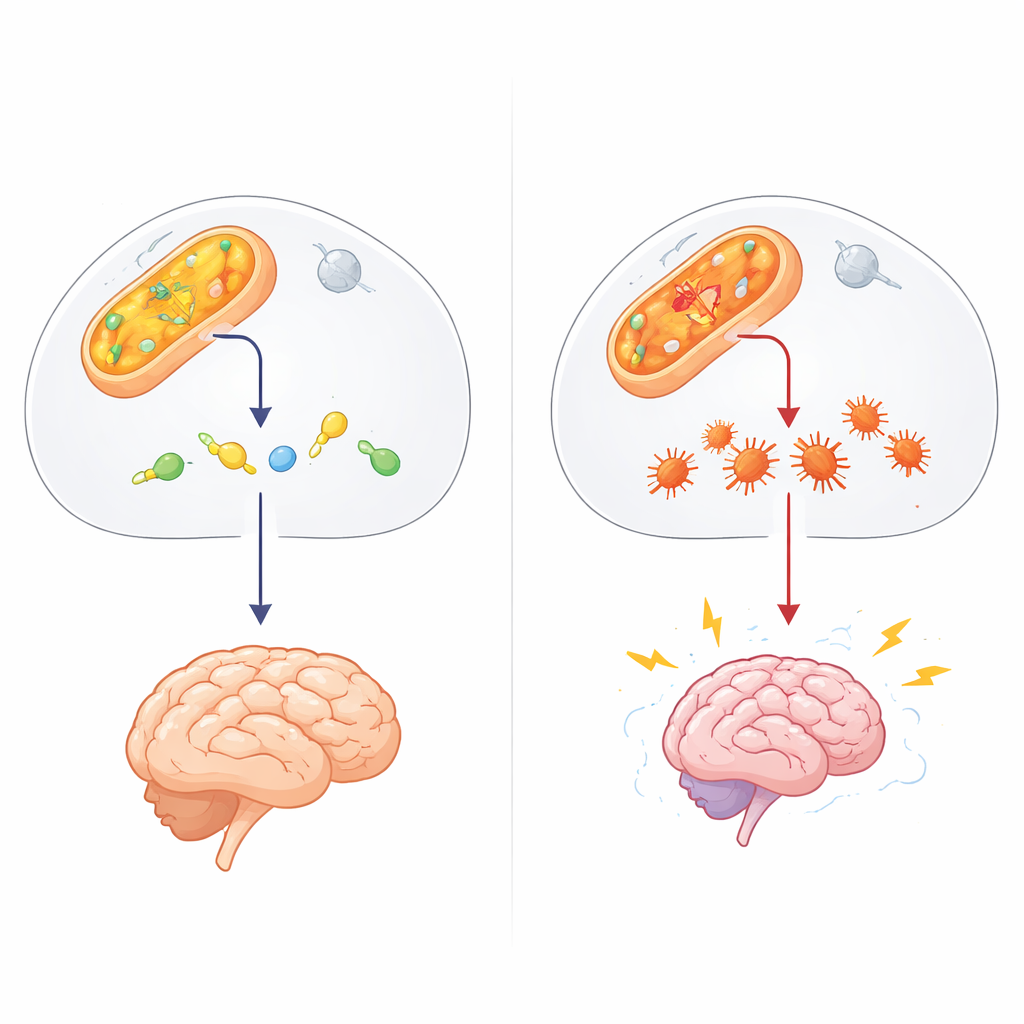
Un gardien méconnu de la chimie cellulaire
Le travail se concentre sur ALDH4A1, membre d’une vaste famille d’enzymes qui détoxifient les aldéhydes — des fragments chimiques hautement réactifs produits par le métabolisme normal, des expositions environnementales et certains médicaments. ALDH4A1 dégrade habituellement un composé lié à l’acide aminé proline, contribuant à maintenir la vitamine B6 active. En l’absence ou lors d’un dysfonctionnement d’ALDH4A1, les personnes développent une hyperprolinémie de type II, une maladie infantile rare caractérisée par des taux très élevés de proline, des crises et un retard de développement. Jusqu’à présent, la plupart des explications mettaient en cause l’excès de proline et une carence en vitamine B6. Pourtant ces hypothèses n’expliquent pas totalement les patients qui ne s’améliorent pas sous supplémentation vitaminique ni les différences étonnantes entre des troubles apparentés.
Une molécule toxique sous les projecteurs
Les auteurs ont suspecté un autre acteur : le 4‑hydroxynonénal (4‑HNE), un aldéhyde adhésif formé lorsque les lipides des membranes cellulaires sont endommagés par le stress oxydatif, par exemple lors des crises. Le 4‑HNE peut se lier aux protéines et à l’ADN, provoquant leur mauvais repliement et leur agrégation. Dans des expériences en éprouvette, l’équipe a montré qu’ALDH4A1 peut directement dégrader le 4‑HNE et, contrairement à une enzyme d’aldéhyde mieux connue, ALDH2, le fait sans être inactivée par le toxique qu’elle tente d’éliminer. Lorsque les chercheurs ont affaibli ALDH4A1 dans des cellules de type nerveux, ces cellules sont devenues beaucoup plus vulnérables au 4‑HNE : elles mouraient plus facilement et accumulaient davantage d’agrégats protéiques, suggérant qu’ALDH4A1 agit normalement comme une protection contre cette molécule dommageable.
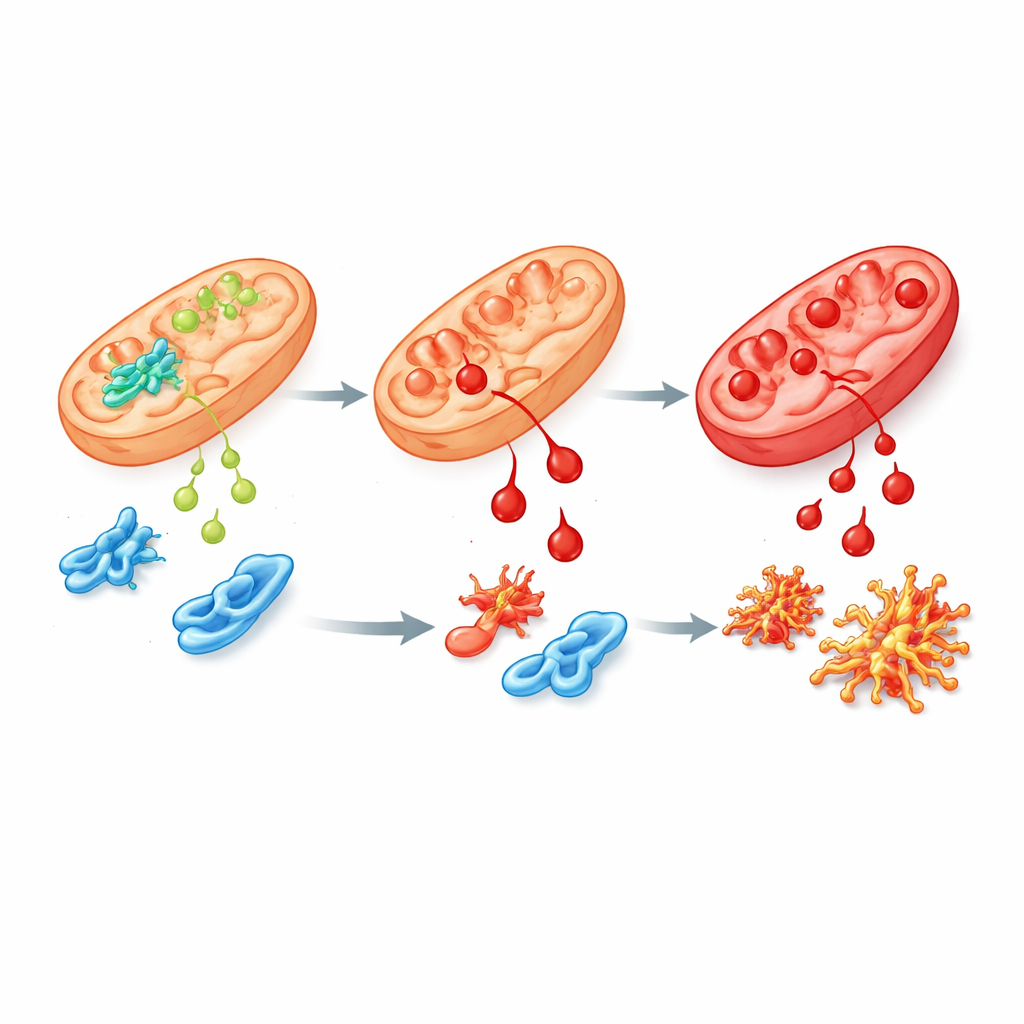
Une variante défectueuse et ses effets en cascade
Pour reproduire plus fidèlement la maladie humaine, l’équipe a modifié des cellules souches humaines et des souris pour porter la mutation la plus fréquente des patients sur ALDH4A1, appelée S352L. Cette altération rend l’enzyme à la fois moins active et très instable, de sorte qu’il n’en reste que peu ou rien dans les cellules. Dans les cellules souches et dans leurs cellules nerveuses et de soutien cultivées en laboratoire, les taux de proline augmentaient fortement, comme observé chez les patients. Ces cellules étaient également beaucoup plus sensibles au 4‑HNE et accumulaient plus rapidement des « cicatrices » chimiques provenant de celui‑ci. L’analyse des gènes activés ou réprimés a révélé de larges perturbations dans les réseaux qui pilotent la croissance cérébrale, gèrent le stress oxydatif et relient la vitamine B6 aux protéines. Plusieurs protéines impliquées dans le métabolisme de la proline et des polyamines, y compris la spermine synthase, étaient réduites, suggérant une perturbation plus vaste de la chimie cérébrale.
Des cellules à l’animal entier
La version murine de la mutation S352L a raconté une histoire semblable au niveau de l’organisme entier. Les souris porteuses de deux copies du gène défectueux présentaient des taux de proline extrêmement élevés dans le sang, l’urine et le cerveau, tout comme les enfants affectés. Beaucoup de ces animaux n’atteignaient pas l’âge adulte, et les portées issues de parents porteurs étaient plus petites que prévu, suggérant que la mutation est délétère dès la vie prénatale. Les souris survivantes étaient plus petites, avaient un cerveau plus léger et montraient des modifications de la composition cellulaire du cerveau, avec moins d’astrocytes par rapport aux neurones. Les astrocytes aident normalement à contrôler l’environnement chimique autour des cellules nerveuses, notamment en gérant des acides aminés comme le glutamate et la proline. Les résultats suggèrent que lorsqu’ALDH4A1 est défectueuse, les astrocytes pourraient libérer un excès de proline et ne pas réussir à protéger les neurones d’une surcharge en aldéhydes, préparant ainsi le terrain aux crises et au mauvais développement cérébral.
Pourquoi cela dépasse une maladie rare
Ensemble, l’étude reconstruit la déficience en ALDH4A1 comme étant plus qu’un simple problème de proline. La perte de cette enzyme amplifie l’impact d’aldéhydes toxiques tels que le 4‑HNE, favorise l’agrégation protéique et reprogramme des programmes génétiques qui façonnent le cerveau en croissance. Cela pourrait expliquer pourquoi la thérapie standard par vitamine B6 est souvent insuffisante et pourquoi les patients conservent des difficultés cognitives. Le travail soulève aussi la possibilité que des variantes plus légères d’ALDH4A1, relativement fréquentes dans la population, augmentent discrètement le risque de troubles liés au stress oxydatif et à l’accumulation protéique, comme certaines formes d’épilepsie, les traumatismes crâniens, l’accident vasculaire cérébral ou même la maladie d’Alzheimer. En cartographiant ces voies, les auteurs soutiennent que l’on peut commencer à concevoir des thérapies visant à stabiliser ou augmenter ALDH4A1, ou à cibler les changements métaboliques et d’expression génétique en aval qu’elle contrôle.
Citation: Kraemer, B.R., Heo, G., Chen, CH. et al. Increased susceptibility to 4-HNE-induced toxicity and impaired development in a model of ALDH4A1-deficient pediatric epilepsy carrying the S352L variant. Commun Biol 9, 597 (2026). https://doi.org/10.1038/s42003-026-09845-y
Mots-clés: épilepsie pédiatrique, aldéhydes toxiques, enzymes mitochondriales, développement cérébral, agrégation protéique