Clear Sky Science · en
Increased susceptibility to 4-HNE-induced toxicity and impaired development in a model of ALDH4A1-deficient pediatric epilepsy carrying the S352L variant
When a Cell’s Cleanup Crew Fails
Some children develop hard-to-treat epilepsy and lifelong learning difficulties for reasons that are still mysterious. This study looks at one such cause: rare changes in a single enzyme inside our cells’ power stations, the mitochondria. By exploring how this enzyme normally helps clear toxic by-products and what happens when it fails, the researchers uncover new clues to why seizures start, why the brain may not develop properly, and how we might design better treatments.
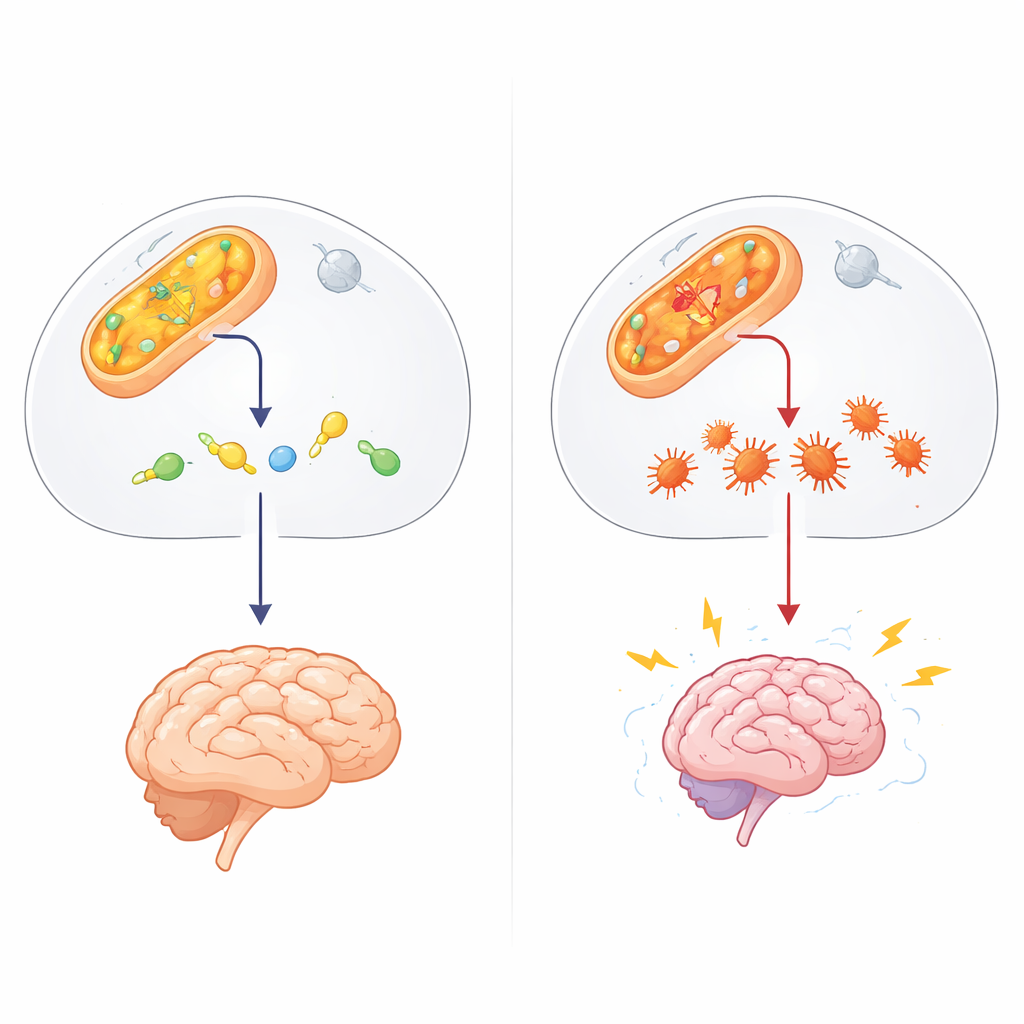
A Hidden Guardian in Cell Chemistry
The work centers on ALDH4A1, part of a large family of enzymes that detoxify aldehydes—highly reactive chemical scraps produced by normal metabolism, environmental exposures, and some medicines. ALDH4A1 usually breaks down a compound linked to the amino acid proline, helping keep vitamin B6 active. When ALDH4A1 is missing or faulty, people develop Hyperprolinemia Type II, a rare childhood disease marked by very high proline levels, seizures, and developmental delay. Until now, most explanations blamed excess proline and a shortage of vitamin B6. Yet those ideas could not fully account for patients who do not improve with vitamin supplements or for puzzling differences between related disorders.
A Toxic Molecule in the Spotlight
The authors suspected another player: 4-hydroxynonenal (4-HNE), a sticky aldehyde formed when fats in cell membranes are damaged by oxidative stress, such as during seizures. 4-HNE can latch onto proteins and DNA, causing them to misfold and clump. In test-tube experiments, the team showed that ALDH4A1 can directly break down 4-HNE, and unlike a better-known aldehyde enzyme, ALDH2, it does so without being shut down by the very toxin it is trying to remove. When the researchers weakened ALDH4A1 in nerve-like cells, those cells became far more vulnerable to 4-HNE: they died more readily and accumulated more protein aggregates, suggesting that ALDH4A1 normally acts as a shield against this damaging molecule.
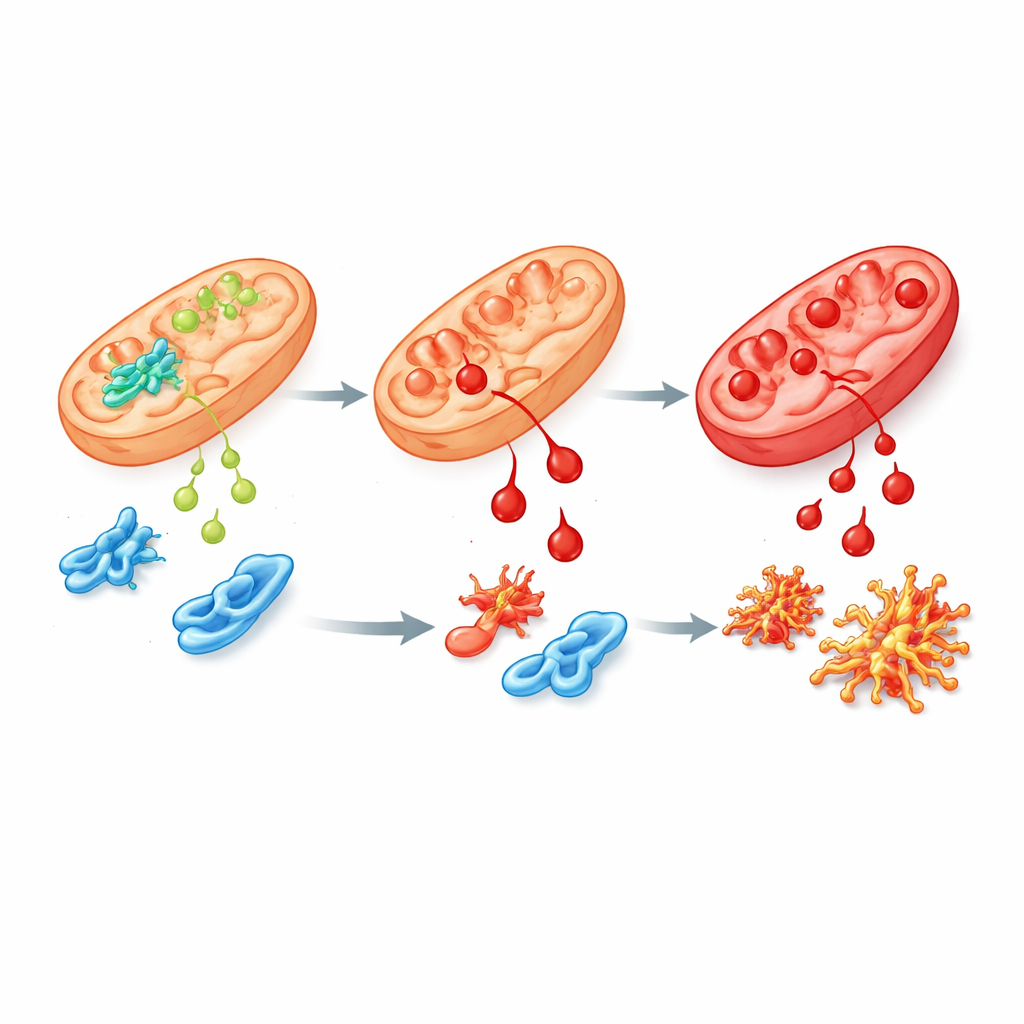
A Faulty Variant and Its Ripple Effects
To mimic the human disease more closely, the team engineered human stem cells and mice to carry the most common patient mutation in ALDH4A1, called S352L. This change makes the enzyme both less active and highly unstable, so little or none of it remains in cells. In stem cells and in their lab-grown nerve and support cells, proline levels rose sharply, as seen in patients. These cells were also far more sensitive to 4-HNE and accumulated chemical “scars” from it more quickly. When the researchers examined which genes were switched on or off, they found broad disturbances in networks that guide brain growth, handle oxidative stress, and connect vitamin B6 to proteins. Several proteins involved in proline and polyamine metabolism, including spermine synthase, were reduced, hinting at wider disruption of brain chemistry.
From Cells to Whole Animals
The mouse version of the S352L mutation told a similar story at the level of the whole body. Mice with two copies of the faulty gene had extremely high proline in blood, urine, and brain, just like affected children. Many of these animals did not survive to adulthood, and litters from carrier parents were smaller than expected, suggesting that the mutation is harmful even before birth. Surviving mice were smaller, had lighter brains, and showed shifts in brain cell makeup, with fewer astrocytes relative to neurons. Astrocytes normally help control the chemical environment around nerve cells, including managing amino acids like glutamate and proline. The findings suggest that when ALDH4A1 is defective, astrocytes may release extra proline and fail to protect neurons from aldehyde overload, setting the stage for seizures and poor brain development.
Why This Matters Beyond a Rare Disease
Taken together, the study recasts ALDH4A1 deficiency as more than a simple proline problem. Loss of this enzyme amplifies the impact of toxic aldehydes such as 4-HNE, drives protein aggregation, and rewires gene programs that shape the growing brain. This could explain why standard vitamin B6 therapy often falls short and why patients continue to struggle with cognition. The work also raises the possibility that milder ALDH4A1 variants, which are relatively common in the population, might quietly increase risk for conditions linked to oxidative stress and protein buildup, such as certain epilepsies, traumatic brain injury, stroke, or even Alzheimer’s disease. By mapping these pathways, the authors argue, we can begin to design therapies that stabilize or boost ALDH4A1, or that target the downstream metabolic and gene-expression changes it controls.
Citation: Kraemer, B.R., Heo, G., Chen, CH. et al. Increased susceptibility to 4-HNE-induced toxicity and impaired development in a model of ALDH4A1-deficient pediatric epilepsy carrying the S352L variant. Commun Biol 9, 597 (2026). https://doi.org/10.1038/s42003-026-09845-y
Keywords: pediatric epilepsy, toxic aldehydes, mitochondrial enzymes, brain development, protein aggregation