Clear Sky Science · sv
NuConf: ett rotamerbibliotek för DNA och RNA och dess implementering i proteindesignprogrammet MUMBO
Varför det spelar roll att omformera DNA och RNA på en dator
Utvecklingen av nya proteiner med datorverktyg har tagit stora kliv framåt de senaste åren, men DNA och RNA har till stor del hamnat på efterkälken. Strukturerna för dessa genetiska molekyler är mindre väl dokumenterade, vilket gör det svårt för artificiell intelligens att lära sig hur de böjer sig, vrider sig och interagerar med proteiner. Denna studie introducerar NuConf, ett nytt sätt att representera formerna hos DNA- och RNA-byggstenar så att befintlig proteindesignmjukvara också kan designa nukleinsyror och deras kontaktytor med proteiner. 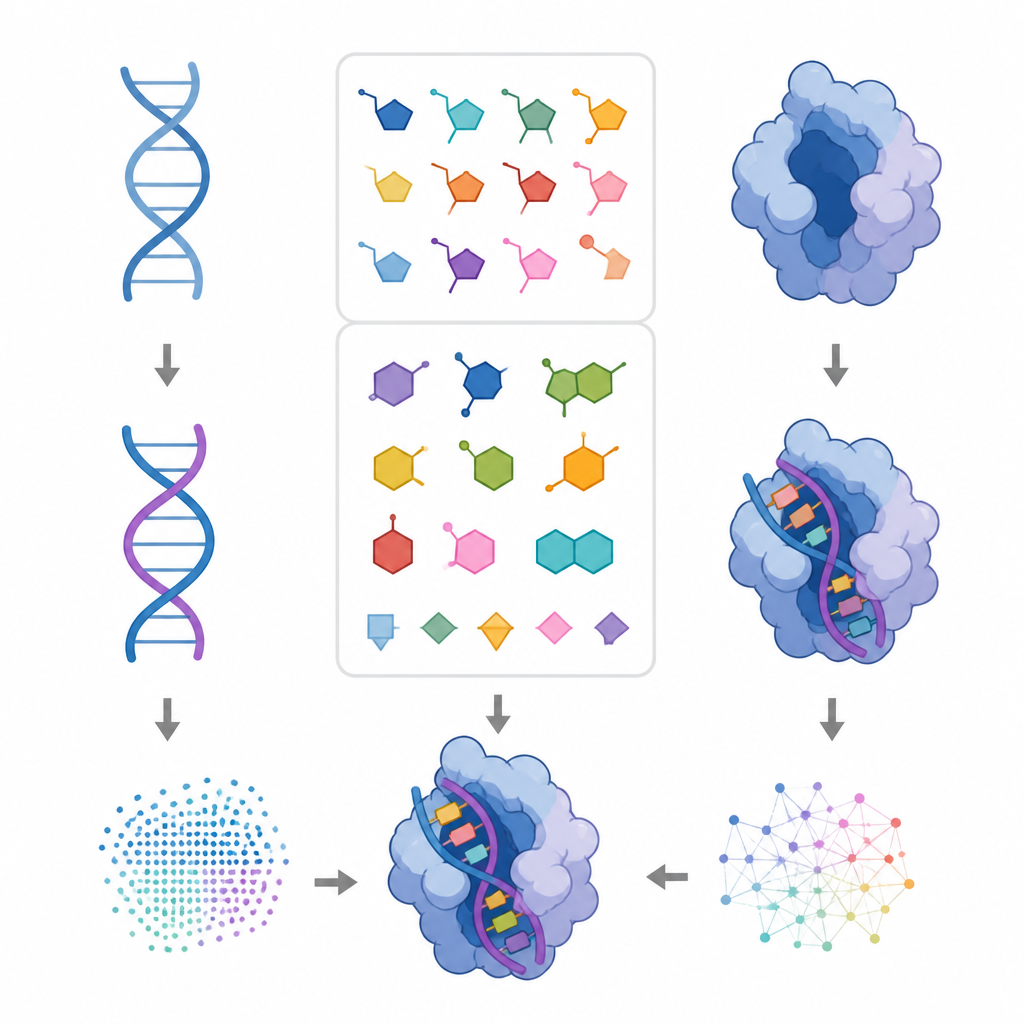
Hur designers vanligtvis hanterar flexibla sidodelar
När forskare designar proteiner på en dator utforskar de inte varje möjlig rörelse i varje sidokedja atom för atom. Istället använder de ”rotamerbibliotek”, samlingar av vanliga sidokedjeformer destillerade från tusentals kända strukturer. Program som MUMBO placerar dessa former på en fixerad proteinryggrad och använder energiuträkningar för att avgöra vilken kombination som passar bäst. Hittills saknades ett motsvarande, praktiskt bibliotek för sidodelarna hos DNA- och RNA-baser, särskilt ett som låter samma program behandla proteiner, DNA och RNA på lika villkor.
Kartläggning av DNA:s och RNA:s favoritformer
Författarna började med att granska mer än 175 000 nukleotider hämtade från högkvalitativa kristallstrukturer av DNA, RNA och deras komplex med proteiner. För varje nukleotid mätte de två nyckelvinklar: en som fångar hur sockerringens puckling ser ut, och en som beskriver hur den bundna basen är roterad i förhållande till den ringen. De fann att sockrarna föredrar två breda familjer av former, en mer typisk för DNA och en för RNA, och att dessa sockermönster starkt hänger samman med hur basen är orienterad. Med andra ord är ryggradens hållning och basens riktning inte oberoende; de rör sig synkront i karaktäristiska mönster.
Att omvandla strukturella mönster till ett praktiskt bibliotek
För att göra dessa mönster användbara i ett designprogram tillämpade teamet statistiska modeller som bryter ner komplicerade vinkel‑fördelningar i ett litet antal distinkta toppar, där varje topp representerar en ofta använd basorientering. För varje bas i DNA och RNA, och för varje bred sockertyp, definierade de tre till sex föredragna orienteringar, tillsammans med hur ofta varje förekommer. Denna samling, kallad NuConf, fungerar som en nukleosid ”formkatalog” knuten till den lokala ryggradens hållning. De skapade också en enklare reservversion med färre former, som byter bort en del detalj mot lägre beräkningskostnad. 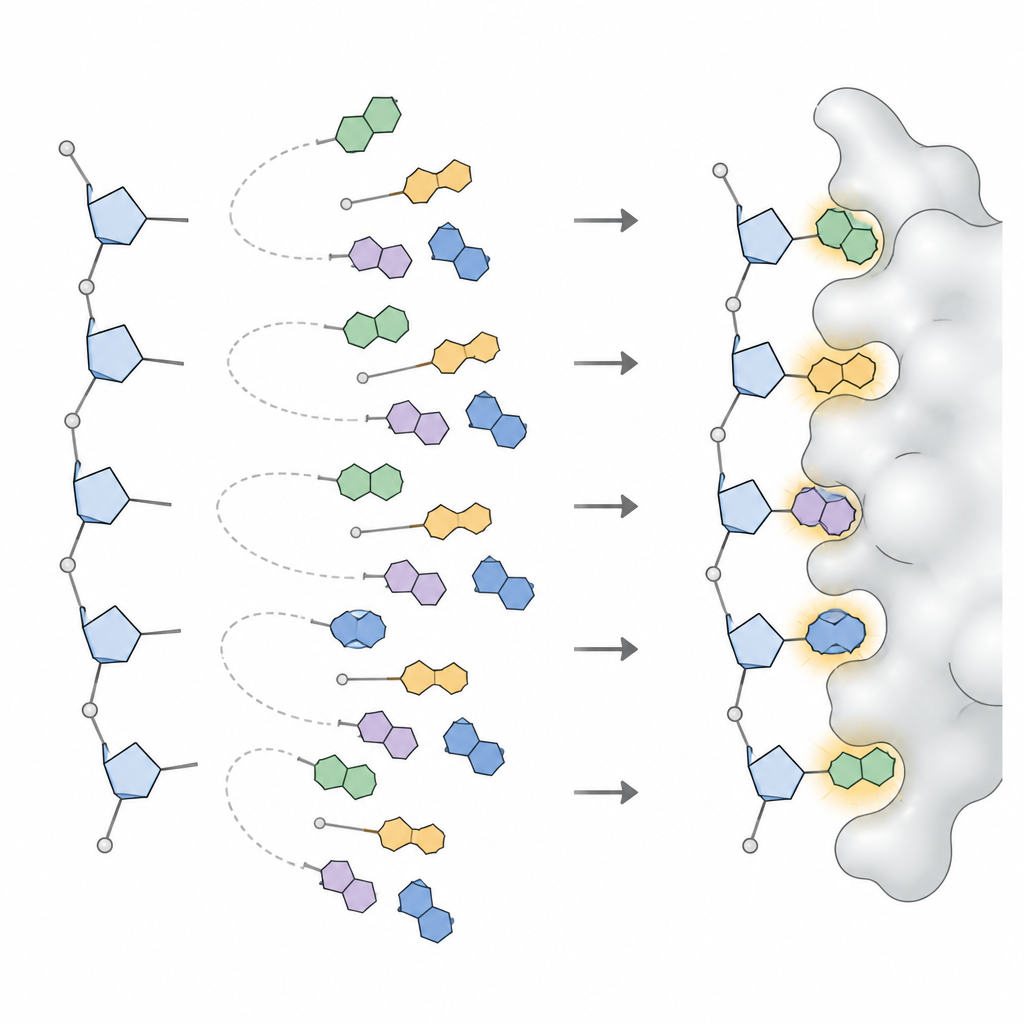
Testa om de nya formerna verkligen fungerar
Forskarna byggde sedan in dessa former i MUMBO och ställde två frågor: kunde programmet, givet en fixerad ryggrad, rekonstruera de ursprungliga baspositionerna som sågs i verkliga strukturer, och kunde det välja bra former och sekvenser enbart utifrån energipoäng, utan att få svaret i förväg? I stora testset innehållande tiotusentals nukleotider reproducerade NuConf‑biblioteket baspositioner med noggrannhet jämförbar med, och ibland bättre än, de standardbibliotek som används för proteinsidokedjor. När programmet var tvunget att välja former enbart utifrån energi överträffade NuConf fortfarande ett enklare bibliotek och konkurrerande nukleinsyraverktyg, samtidigt som det fångade viktiga basparnings‑ och stackningskontakter i både fria nukleinsyror och protein–nukleinsyra‑komplex.
Vad detta betyder för framtida molekyldesign
För icke‑specialister är huvudpoängen att författarna har gett datorstödd design ett nytt, gemensamt språk för både proteiner och nukleinsyror. NuConf gör det möjligt för befintlig proteindesignmjukvara att pålitligt placera och välja DNA‑ och RNA‑baser längs en given ryggrad och vid protein‑kontaktställen. Detta ersätter inte moderna AI‑metoder, men fyller ett viktigt gap när träningsdata är knappa eller när finmaskiga fysikaliska interaktioner måste utvärderas. På längre sikt skulle denna typ av verktyg kunna hjälpa forskare att designa mer precisa genregulatorer, RNA‑switchar och hybrida protein–nukleinsyra‑maskiner helt in silico innan de byggs i laboratoriet.
Citering: Makarova, M.O., Stiebritz, M.T., Basturk, D. et al. NuConf: a rotamer library for DNA and RNA and its implementation in the protein design software MUMBO. Sci Rep 16, 16281 (2026). https://doi.org/10.1038/s41598-026-52380-3
Nyckelord: design av nukleinsyror, rotamerbibliotek, protein–DNA-interaktioner, RNA-modellering, beräkningsbaserad strukturell biologi