Clear Sky Science · en
NuConf: a rotamer library for DNA and RNA and its implementation in the protein design software MUMBO
Why reshaping DNA and RNA on a computer matters
Designing new proteins with computer tools has leapt forward in recent years, but DNA and RNA have largely been left behind. The structures of these genetic molecules are more sparsely documented, which makes it hard for artificial intelligence to learn how they bend, twist, and interact with proteins. This study introduces NuConf, a new way to represent the shapes of DNA and RNA building blocks so that existing protein design software can also design nucleic acids and their contact surfaces with proteins. 
How designers usually handle flexible side parts
When scientists design proteins on a computer, they do not explore every possible wiggle of each side chain atom by atom. Instead, they use “rotamer libraries,” collections of common side chain shapes distilled from thousands of known structures. Programs like MUMBO place these shapes onto a fixed protein backbone and use energy calculations to decide which combination fits best. Until now, a comparable, practical library for the side parts of DNA and RNA bases was missing, especially one that would let the same program treat proteins, DNA, and RNA on equal footing.
Mapping the favorite shapes of DNA and RNA
The authors began by examining more than 175,000 nucleotides taken from high‑quality crystal structures of DNA, RNA, and their complexes with proteins. For each nucleotide, they measured two key angles: one that captures how the sugar ring of the backbone puckers, and one that describes how the attached base is rotated relative to that sugar. They found that sugars favor two broad families of shapes, one more typical of DNA and one of RNA, and that these sugar shapes are strongly linked to how the base is oriented. In other words, the backbone posture and base direction are not independent; they move in step in characteristic ways.
Turning structural patterns into a practical library
To turn these patterns into something a design program can use, the team applied statistical models that break complicated angle distributions into a small set of distinct peaks, each peak representing a frequently used base orientation. For every type of base in DNA and RNA, and for each broad sugar shape, they defined three to six preferred orientations, along with how often each occurs. This collection, called NuConf, acts as a nucleoside “shape catalog” tied to the local backbone posture. They also created a simpler backup version with fewer shapes, which trades some detail for lower computing cost. 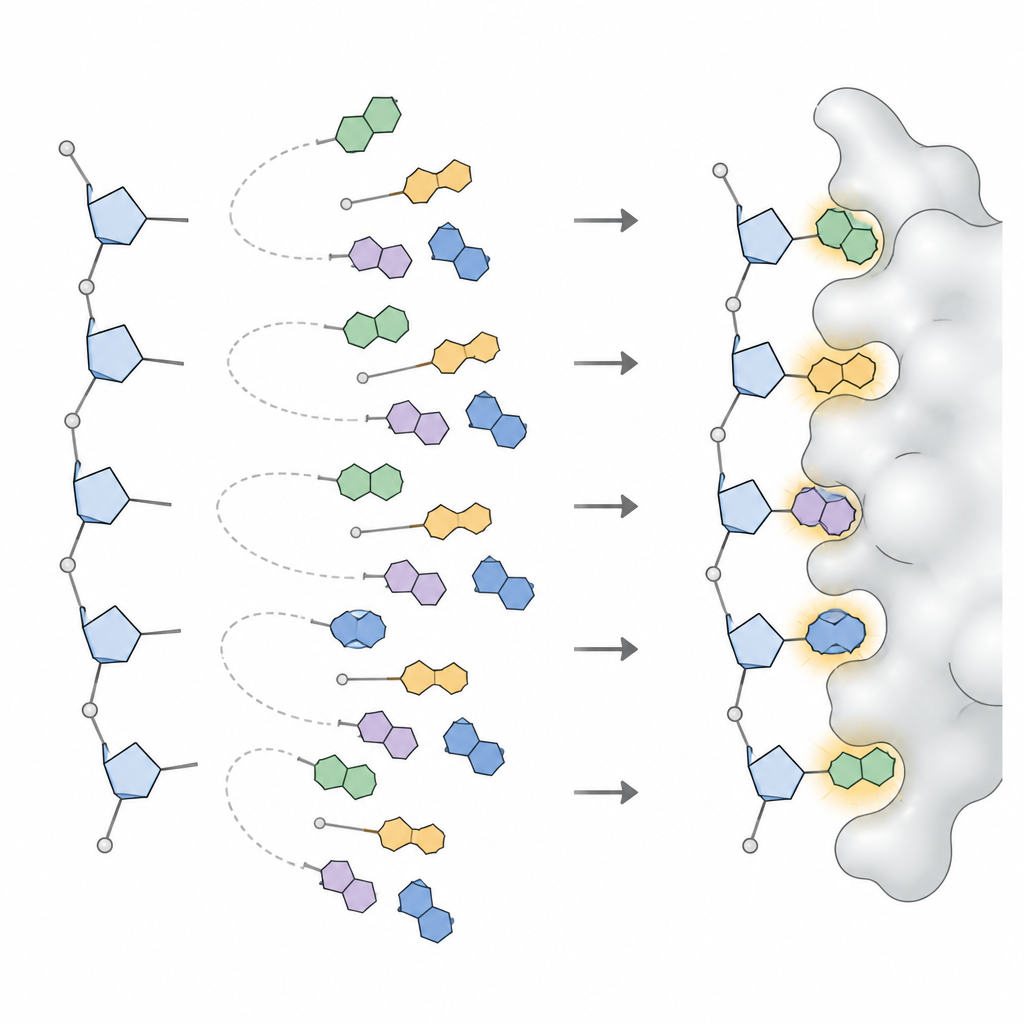
Testing whether the new shapes really work
The researchers then built these shapes into MUMBO and asked two questions: could the program, given a fixed backbone, reconstruct the original base positions seen in real structures, and could it pick good shapes and sequences using only energy scores, without being told the answer? In large test sets containing tens of thousands of nucleotides, the NuConf library reproduced base positions with accuracy comparable to, and sometimes better than, that of standard libraries used for protein side chains. When the program had to choose shapes purely by energy, NuConf still outperformed a simpler library and competing nucleic acid tools, while capturing key base‑pairing and stacking contacts in both free nucleic acids and protein‑nucleic acid complexes.
What this means for future molecular design
For non‑specialists, the main takeaway is that the authors have given computer‑aided design a new, shared language for both proteins and nucleic acids. NuConf lets existing protein design software reliably place and choose DNA and RNA bases along a given backbone and at protein contact sites. This does not replace modern AI methods, but it fills an important gap when training data are scarce or when fine‑grained physical interactions must be evaluated. In the long run, this kind of tool could help researchers design more precise gene regulators, RNA switches, and hybrid protein‑nucleic acid machines entirely in silico before they are built in the lab.
Citation: Makarova, M.O., Stiebritz, M.T., Basturk, D. et al. NuConf: a rotamer library for DNA and RNA and its implementation in the protein design software MUMBO. Sci Rep 16, 16281 (2026). https://doi.org/10.1038/s41598-026-52380-3
Keywords: nucleic acid design, rotamer library, protein–DNA interactions, RNA modeling, computational structural biology