Clear Sky Science · pt
NuConf: uma biblioteca de rotâmeros para DNA e RNA e sua implementação no software de design de proteínas MUMBO
Por que remodelar DNA e RNA no computador importa
Projetar novas proteínas com ferramentas computacionais avançou muito nos últimos anos, mas DNA e RNA ficaram em grande parte para trás. As estruturas dessas moléculas genéticas estão menos documentadas, o que dificulta o aprendizado de como elas se dobram, torcem e interagem com proteínas por métodos de inteligência artificial. Este estudo apresenta o NuConf, uma nova forma de representar as geometrias dos blocos construtores de DNA e RNA para que softwares existentes de design de proteínas também possam projetar ácidos nucleicos e suas superfícies de contato com proteínas. 
Como os projetistas geralmente lidam com partes laterais flexíveis
Quando cientistas projetam proteínas no computador, eles não exploram cada possível movimento de cada cadeia lateral átomo a átomo. Em vez disso, usam “bibliotecas de rotâmeros”, coleções de formas comuns de cadeias laterais extraídas de milhares de estruturas conhecidas. Programas como o MUMBO posicionam essas formas sobre um esqueleto proteico fixo e usam cálculos de energia para decidir qual combinação se ajusta melhor. Até agora, faltava uma biblioteca prática e comparável para as partes laterais das bases de DNA e RNA, especialmente uma que permitisse ao mesmo programa tratar proteínas, DNA e RNA em pé de igualdade.
Mapeando as formas preferidas de DNA e RNA
Os autores começaram examinando mais de 175.000 nucleotídeos extraídos de estruturas cristalográficas de alta qualidade de DNA, RNA e seus complexos com proteínas. Para cada nucleotídeo, mediram dois ângulos chave: um que captura como o anel de açúcar da coluna vertebral se projeta, e outro que descreve como a base ligada está rotacionada em relação a esse açúcar. Concluíram que os açúcares favorecem duas grandes famílias de conformações, uma mais típica do DNA e outra do RNA, e que essas conformações do açúcar estão fortemente ligadas à orientação da base. Em outras palavras, a postura do esqueleto e a direção da base não são independentes; elas se movem em conjunto de maneiras características.
Transformando padrões estruturais em uma biblioteca prática
Para converter esses padrões em algo utilizável por um programa de design, a equipe aplicou modelos estatísticos que decomponham distribuições angulares complexas em um pequeno conjunto de picos distintos, cada pico representando uma orientação de base frequentemente usada. Para cada tipo de base em DNA e RNA, e para cada forma ampla de açúcar, definiram de três a seis orientações preferidas, juntamente com a frequência de ocorrência de cada uma. Essa coleção, chamada NuConf, funciona como um “catálogo de formas” de nucleosídeos atrelado à postura local do esqueleto. Eles também criaram uma versão simplificada com menos formas, que sacrifica um pouco de detalhe em favor de menor custo computacional. 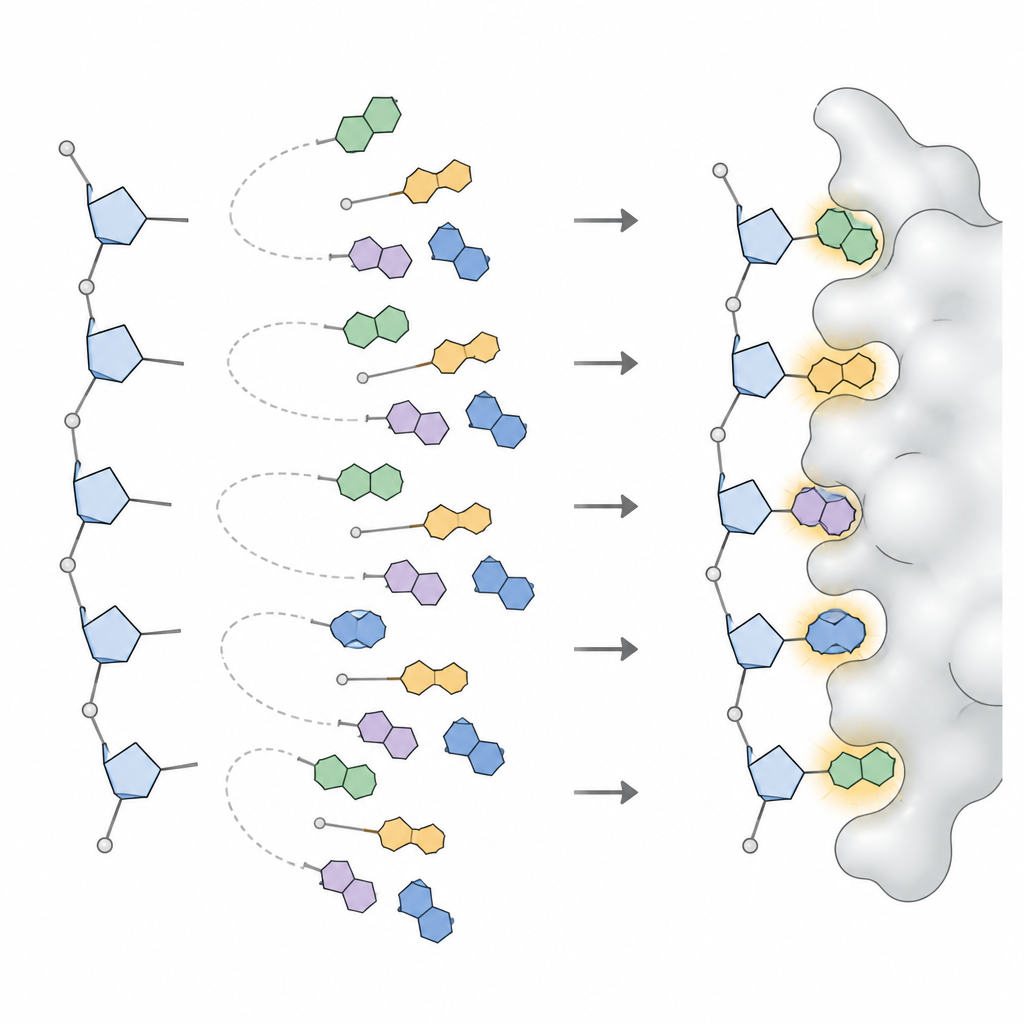
Testando se as novas formas realmente funcionam
Os pesquisadores então incorporaram essas formas ao MUMBO e fizeram duas perguntas: o programa, dado um esqueleto fixo, poderia reconstruir as posições das bases observadas em estruturas reais, e poderia escolher boas formas e sequências usando apenas escores de energia, sem ser informado da resposta? Em grandes conjuntos de teste contendo dezenas de milhares de nucleotídeos, a biblioteca NuConf reproduziu posições de bases com precisão comparável e, às vezes, superior à de bibliotecas padrão usadas para cadeias laterais de proteínas. Quando o programa teve que escolher formas puramente por energia, o NuConf ainda superou uma biblioteca mais simples e ferramentas concorrentes de ácidos nucleicos, além de captar contatos chave de pareamento e empilhamento de bases tanto em ácidos nucleicos livres quanto em complexos proteína–ácido nucleico.
O que isso significa para o futuro do design molecular
Para não especialistas, a principal conclusão é que os autores deram ao design assistido por computador uma nova linguagem compartilhada para proteínas e ácidos nucleicos. O NuConf permite que softwares de design de proteínas existentes coloquem e escolham com confiança bases de DNA e RNA ao longo de um esqueleto dado e em sítios de contato com proteínas. Isso não substitui métodos modernos de IA, mas preenche uma lacuna importante quando os dados de treinamento são escassos ou quando interações físicas detalhadas precisam ser avaliadas. A longo prazo, esse tipo de ferramenta pode ajudar pesquisadores a projetar reguladores gênicos mais precisos, chaves de RNA e máquinas híbridas proteína–ácido nucleico inteiramente in silico antes de serem construídas no laboratório.
Citação: Makarova, M.O., Stiebritz, M.T., Basturk, D. et al. NuConf: a rotamer library for DNA and RNA and its implementation in the protein design software MUMBO. Sci Rep 16, 16281 (2026). https://doi.org/10.1038/s41598-026-52380-3
Palavras-chave: design de ácidos nucleicos, biblioteca de rotâmeros, interações proteína–DNA, modelagem de RNA, biologia estrutural computacional