Clear Sky Science · es
NuConf: una biblioteca de rotámeros para ADN y ARN y su implementación en el software de diseño de proteínas MUMBO
Por qué importa remodelar ADN y ARN en el ordenador
El diseño de nuevas proteínas con herramientas computacionales ha avanzado mucho en los últimos años, pero el ADN y el ARN han quedado en gran medida rezagados. Las estructuras de estas moléculas genéticas están menos documentadas, lo que dificulta que la inteligencia artificial aprenda cómo se doblan, giran e interactúan con proteínas. Este estudio presenta NuConf, una nueva forma de representar las geometrías de los bloques constructores del ADN y ARN para que el software de diseño de proteínas existente pueda también diseñar ácidos nucleicos y sus superficies de contacto con proteínas. 
Cómo gestionan normalmente los diseñadores las partes laterales flexibles
Cuando los científicos diseñan proteínas por ordenador, no exploran cada movimiento posible de cada cadena lateral átomo por átomo. En su lugar, emplean «bibliotecas de rotámeros», colecciones de formas comunes de cadenas laterales obtenidas de miles de estructuras conocidas. Programas como MUMBO colocan esas formas sobre un esqueleto proteico fijo y usan cálculos energéticos para decidir qué combinación encaja mejor. Hasta ahora faltaba una biblioteca práctica comparable para las partes laterales de las bases de ADN y ARN, especialmente una que permitiera al mismo programa tratar proteínas, ADN y ARN en igualdad de condiciones.
Cartografiar las formas preferidas de ADN y ARN
Los autores empezaron examinando más de 175.000 nucleótidos extraídos de estructuras cristalográficas de alta calidad de ADN, ARN y sus complejos con proteínas. Para cada nucleótido midieron dos ángulos clave: uno que captura cómo se pandea el anillo de azúcar de la columna vertebral y otro que describe cómo está girada la base con respecto a ese azúcar. Encontraron que los azúcares prefieren dos grandes familias de conformaciones, una más típica del ADN y otra del ARN, y que esas conformaciones del azúcar están fuertemente ligadas a la orientación de la base. En otras palabras, la postura de la columna vertebral y la dirección de la base no son independientes; se desplazan de forma coordinada y característica.
Convertir patrones estructurales en una biblioteca práctica
Para convertir estos patrones en algo que un programa de diseño pueda usar, el equipo aplicó modelos estadísticos que descomponen distribuciones angulares complejas en un pequeño conjunto de picos distintos, cada pico representando una orientación de base frecuentemente usada. Para cada tipo de base en ADN y ARN, y para cada amplia conformación del azúcar, definieron de tres a seis orientaciones preferidas, junto con su frecuencia de aparición. Esta colección, llamada NuConf, actúa como un «catálogo de formas» de nucleósidos ligado a la postura local del esqueleto. También crearon una versión de reserva más sencilla con menos formas, que sacrifica algo de detalle a cambio de menor coste computacional. 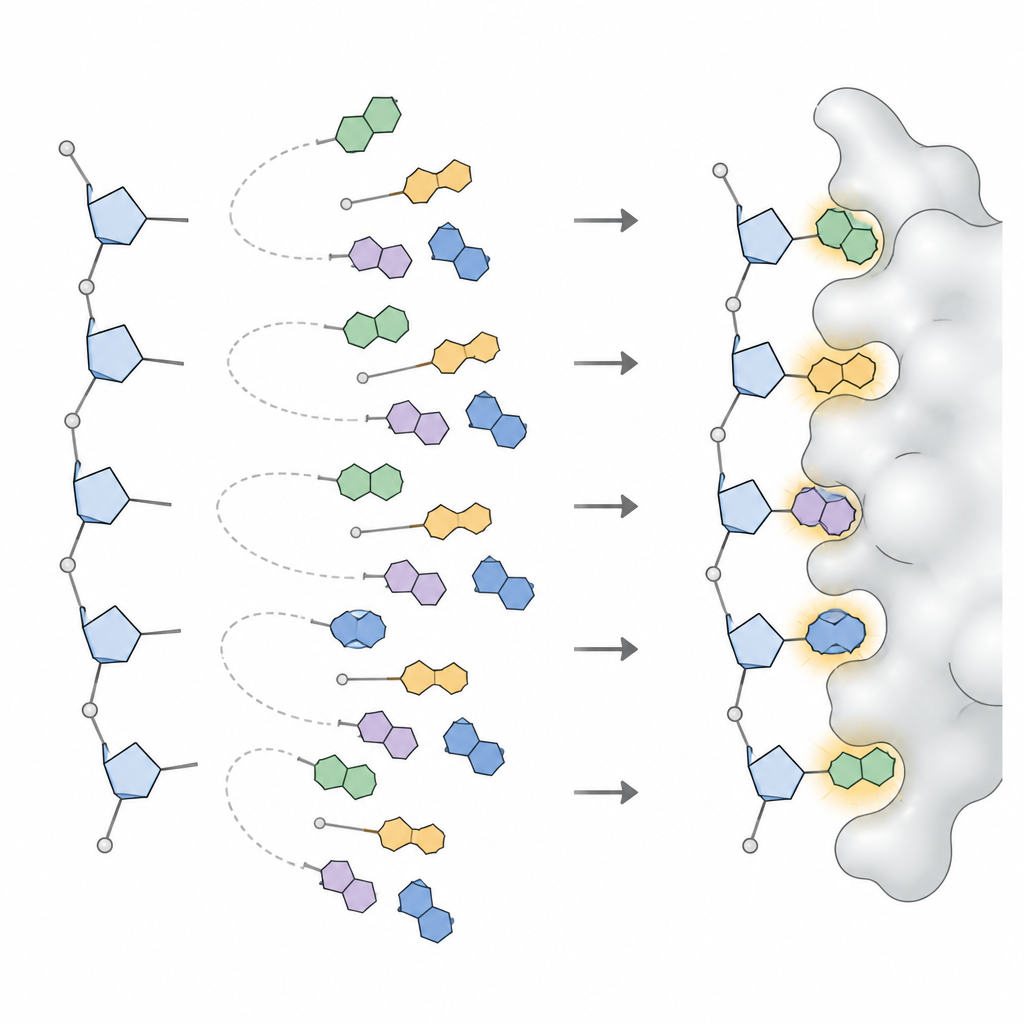
Probar si las nuevas formas funcionan realmente
Los investigadores incorporaron entonces estas formas en MUMBO y formularon dos preguntas: con un esqueleto fijo, ¿podría el programa reconstruir las posiciones originales de las bases observadas en estructuras reales?, y ¿podría elegir buenas formas y secuencias usando solo puntuaciones energéticas, sin que se le indiquen las respuestas? En grandes conjuntos de prueba con decenas de miles de nucleótidos, la biblioteca NuConf reprodujo las posiciones de las bases con una precisión comparable y a veces superior a la de las bibliotecas estándar usadas para cadenas laterales de proteínas. Cuando el programa tuvo que elegir formas únicamente por energía, NuConf siguió superando a una biblioteca más simple y a otras herramientas de ácidos nucleicos competidoras, a la vez que capturaba contactos clave de apareamiento y apilamiento de bases tanto en ácidos nucleicos libres como en complejos proteína‑ácido nucleico.
Qué significa esto para el diseño molecular futuro
Para los no especialistas, la conclusión principal es que los autores han dado al diseño asistido por ordenador un nuevo lenguaje común para proteínas y ácidos nucleicos. NuConf permite que el software de diseño de proteínas existente sitúe y seleccione de forma fiable bases de ADN y ARN a lo largo de un esqueleto dado y en sitios de contacto con proteínas. Esto no reemplaza los métodos modernos de IA, pero llena un hueco importante cuando los datos de entrenamiento son escasos o cuando hay que evaluar interacciones físicas de detalle fino. A largo plazo, este tipo de herramienta podría ayudar a los investigadores a diseñar reguladores génicos más precisos, conmutadores de ARN y máquinas híbridas proteína‑ácido nucleico totalmente in silico antes de construirlos en el laboratorio.
Cita: Makarova, M.O., Stiebritz, M.T., Basturk, D. et al. NuConf: a rotamer library for DNA and RNA and its implementation in the protein design software MUMBO. Sci Rep 16, 16281 (2026). https://doi.org/10.1038/s41598-026-52380-3
Palabras clave: diseño de ácidos nucleicos, biblioteca de rotámeros, interacciones proteína‑ADN, modelado de ARN, biología estructural computacional