Clear Sky Science · de
NuConf: eine Rotamerbibliothek für DNA und RNA und ihre Implementierung in der Protein‑Design‑Software MUMBO
Warum die digitale Umformung von DNA und RNA wichtig ist
Das Design neuer Proteine mit Computerwerkzeugen hat in den letzten Jahren große Fortschritte gemacht, doch DNA und RNA wurden dabei weitgehend vernachlässigt. Die Strukturen dieser genetischen Moleküle sind weniger umfassend dokumentiert, weshalb es für künstliche Intelligenz schwer ist zu lernen, wie sie sich biegen, verdrehen und mit Proteinen interagieren. Diese Studie stellt NuConf vor, eine neue Darstellung der Formen von DNA‑ und RNA‑Bausteinen, damit vorhandene Protein‑Design‑Software auch Nukleinsäuren und ihre Kontaktflächen mit Proteinen entwerfen kann. 
Wie Designer üblicherweise mit flexiblen Seitenbestandteilen umgehen
Wenn Wissenschaftler Proteine am Computer entwerfen, untersuchen sie nicht atomgenau jede mögliche Bewegung jeder Seitenkette. Stattdessen verwenden sie „Rotamerbibliotheken“, Sammlungen typischer Seitenkettenformen, die aus Tausenden bekannter Strukturen extrahiert wurden. Programme wie MUMBO platzieren diese Formen auf einem festen Protein‑Backbone und verwenden Energieberechnungen, um zu entscheiden, welche Kombination am besten passt. Bisher fehlte eine vergleichbare, praktikable Bibliothek für die Seitenbestandteile von DNA‑ und RNA‑Basen, insbesondere eine, die es demselben Programm erlaubt, Proteine, DNA und RNA gleichberechtigt zu behandeln.
Die bevorzugten Formen von DNA und RNA kartieren
Die Autorinnen und Autoren begannen damit, mehr als 175.000 Nukleotide aus hochwertigen Kristallstrukturen von DNA, RNA und ihren Komplexen mit Proteinen zu untersuchen. Für jedes Nukleotid maßen sie zwei zentrale Winkel: einen, der die Puckering‑Form des Zucker‑Rings im Backbone erfasst, und einen, der beschreibt, wie die angehängte Base relativ zu diesem Zucker rotiert ist. Sie stellten fest, dass Zucker zwei grobe Formfamilien bevorzugen, eine typischer für DNA und eine für RNA, und dass diese Zuckerformen stark mit der Orientierung der Base verknüpft sind. Mit anderen Worten: Backbone‑Haltung und Basenrichtung sind nicht unabhängig; sie bewegen sich in charakteristischen Mustern miteinander.
Strukturmuster in eine praktische Bibliothek überführen
Um diese Muster in etwas umzuwandeln, das ein Designprogramm nutzen kann, wandte das Team statistische Modelle an, die komplizierte Winkelverteilungen in eine kleine Anzahl separater Spitzen zerlegen, wobei jede Spitze eine häufig verwendete Basenorientierung repräsentiert. Für jeden Basentyp in DNA und RNA und für jede der breiten Zuckerformen definierten sie drei bis sechs bevorzugte Orientierungen sowie deren Auftretenshäufigkeit. Diese Sammlung, NuConf genannt, fungiert als Nukleosid‑„Formkatalog“, der an die lokale Backbone‑Haltung gebunden ist. Außerdem erstellten sie eine einfachere Backup‑Version mit weniger Formen, die etwas Genauigkeit gegen geringere Rechenkosten eintauscht. 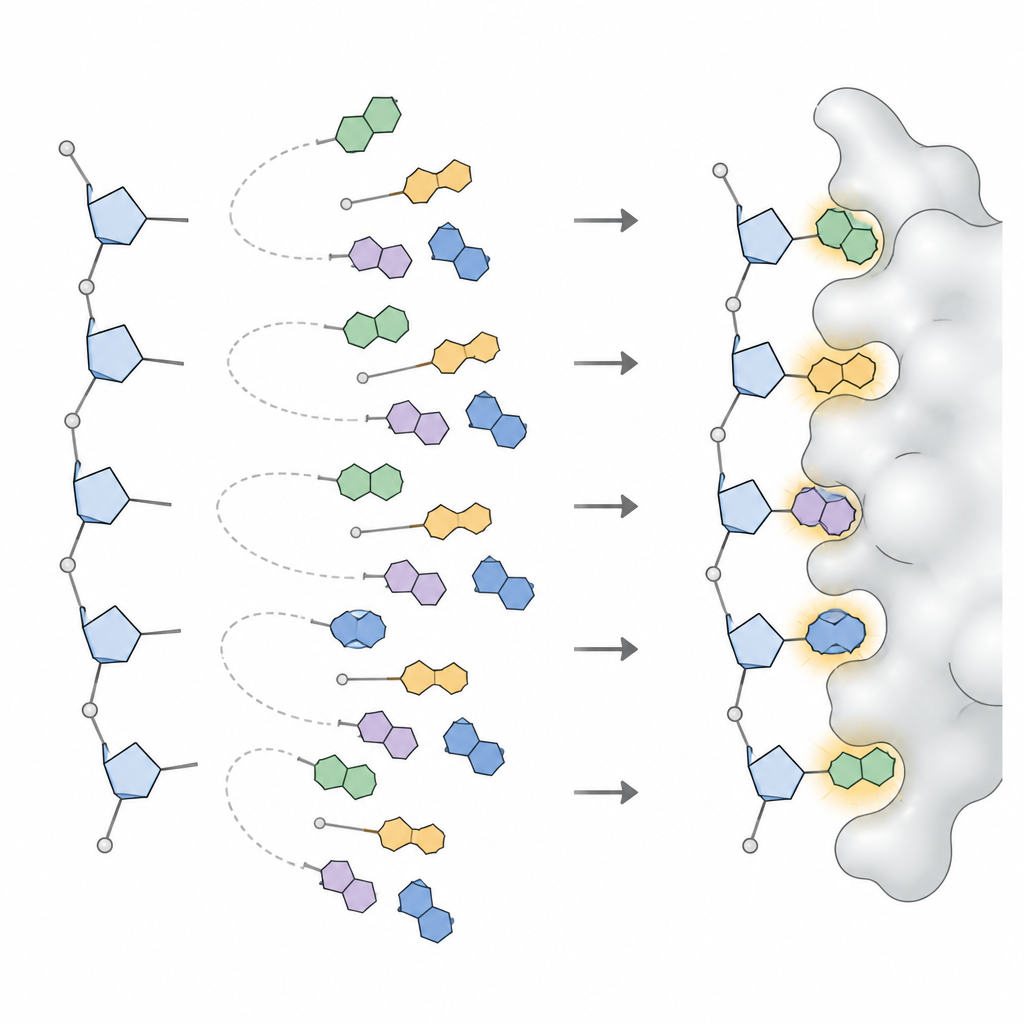
Prüfen, ob die neuen Formen tatsächlich funktionieren
Die Forschenden bauten diese Formen dann in MUMBO ein und stellten zwei Fragen: Konnte das Programm bei festem Backbone die ursprünglichen Basepositionen aus realen Strukturen rekonstruieren, und konnte es gute Formen und Sequenzen allein anhand von Energiewerten auswählen, ohne das Ergebnis vorgegeben zu bekommen? In großen Testsätzen mit Zehntausenden Nukleotiden reproduzierte die NuConf‑Bibliothek die Basepositionen mit einer Genauigkeit, die mit der standardmäßig für Proteineseitenketten verwendeten Bibliotheken vergleichbar war und sie in manchen Fällen übertraf. Wenn das Programm Formen nur anhand von Energie auswählte, übertraf NuConf weiterhin eine einfachere Bibliothek und konkurrierende Nukleinsäure‑Werkzeuge und erfasste dabei wichtige Basenpaarungs‑ und Stapelungskontakte sowohl in freien Nukleinsäuren als auch in Protein‑Nukleinsäure‑Komplexen.
Was das für zukünftiges molekulares Design bedeutet
Für Nichtfachleute ist die wichtigste Erkenntnis, dass die Autorinnen und Autoren dem computerunterstützten Design eine neue, gemeinsame Sprache für Proteine und Nukleinsäuren gegeben haben. NuConf ermöglicht es vorhandener Protein‑Design‑Software, DNA‑ und RNA‑Basen entlang eines gegebenen Backbones und an Protein‑Kontaktstellen zuverlässig zu platzieren und auszuwählen. Dies ersetzt moderne KI‑Methoden nicht, füllt aber eine wichtige Lücke, wenn Trainingsdaten knapp sind oder fein abgestimmte physikalische Wechselwirkungen bewertet werden müssen. Langfristig könnte ein derartiges Werkzeug Forschenden helfen, präzisere Genregulatoren, RNA‑Schalter und hybride Protein‑Nukleinsäure‑Maschinen vollständig in silico zu entwerfen, bevor sie im Labor gebaut werden.
Zitation: Makarova, M.O., Stiebritz, M.T., Basturk, D. et al. NuConf: a rotamer library for DNA and RNA and its implementation in the protein design software MUMBO. Sci Rep 16, 16281 (2026). https://doi.org/10.1038/s41598-026-52380-3
Schlüsselwörter: Design von Nukleinsäuren, Rotamerbibliothek, Protein–DNA‑Wechselwirkungen, RNA‑Modellierung, computationale Strukturbiologie