Clear Sky Science · pl
NuConf: biblioteka rotamerów dla DNA i RNA oraz jej implementacja w oprogramowaniu do projektowania białek MUMBO
Dlaczego modelowanie kształtów DNA i RNA na komputerze ma znaczenie
Projektowanie nowych białek przy użyciu narzędzi komputerowych wykonało duży krok naprzód w ostatnich latach, jednak DNA i RNA w dużej mierze zostały z tyłu. Struktury tych cząsteczek genetycznych są rzadziej udokumentowane, co utrudnia sztucznej inteligencji nauczenie się, jak się wyginają, skręcają i oddziałują z białkami. W tym badaniu przedstawiono NuConf — nowy sposób reprezentacji kształtów fragmentów DNA i RNA, dzięki któremu istniejące oprogramowanie do projektowania białek może także projektować kwasy nukleinowe i ich powierzchnie kontaktowe z białkami. 
Jak projektanci zwykle radzą sobie z elastycznymi fragmentami bocznymi
Kiedy naukowcy projektują białka na komputerze, nie eksplorują każdej możliwej konfiguracji każdego atomu łańcucha bocznego atom po atomie. Zamiast tego używają „bibliotek rotamerów” — zbiorów powszechnych kształtów łańcuchów bocznych wyprowadzonych z tysięcy znanych struktur. Programy takie jak MUMBO umieszczają te kształty na ustalonym szkielecie białkowym i używają obliczeń energii, aby zdecydować, które kombinacje pasują najlepiej. Do tej pory brakowało porównywalnej, praktycznej biblioteki dla fragmentów bocznych zasad DNA i RNA, szczególnie takiej, która pozwoliłaby temu samemu programowi traktować białka, DNA i RNA na równych zasadach.
Mapowanie ulubionych kształtów DNA i RNA
Autorzy zaczęli od zbadania ponad 175 000 nukleotydów pochodzących z wysokiej jakości struktur krystalicznych DNA, RNA i ich kompleksów z białkami. Dla każdego nukleotydu zmierzyli dwa kluczowe kąty: jeden odzwierciedlający kształt „wypychania” pierścienia cukrowego w szkielecie, oraz drugi opisujący, jak przyłączona zasada jest obrócona względem tego cukru. Odkryli, że cukry preferują dwie główne rodziny kształtów — jedną bardziej typową dla DNA, drugą dla RNA — i że te kształty cukrowe są silnie powiązane z orientacją zasady. Innymi słowy, postura szkieletu i kierunek zasady nie są niezależne; poruszają się w charakterystyczny sposób.
Przekształcanie wzorców strukturalnych w praktyczną bibliotekę
Aby przekształcić te wzorce w coś, czego może używać program do projektowania, zespół zastosował modele statystyczne rozbijające złożone rozkłady kątów na niewielki zestaw odrębnych pików, z których każdy reprezentuje często występującą orientację zasady. Dla każdego typu zasady w DNA i RNA oraz dla każdej szerokiej rodziny kształtów cukru zdefiniowano od trzech do sześciu preferowanych orientacji, wraz z częstością ich występowania. Ten zbiór, nazwany NuConf, działa jak „katalog kształtów” nukleozydów powiązany z lokalną postawą szkieletu. Użyto także uproszczonej, zapasowej wersji z mniejszą liczbą kształtów, która oddaje część szczegółów na rzecz niższego kosztu obliczeniowego. 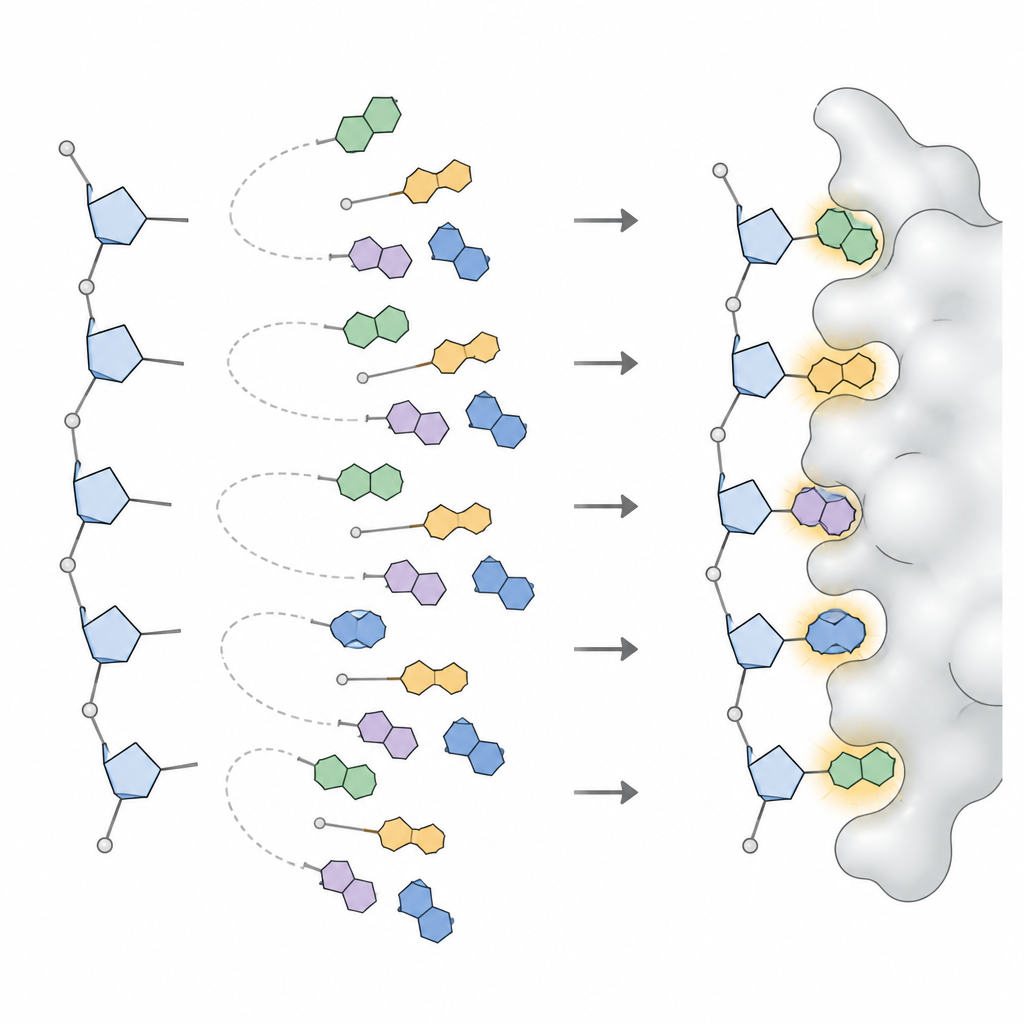
Testowanie, czy nowe kształty rzeczywiście działają
Następnie badacze zaimplementowali te kształty w MUMBO i zadali dwa pytania: czy program, mając ustalony szkielet, potrafi odtworzyć pierwotne pozycje zasad widziane w rzeczywistych strukturach, oraz czy potrafi wybrać dobre kształty i sekwencje używając wyłącznie ocen energetycznych, bez podpowiedzi? W dużych zestawach testowych zawierających dziesiątki tysięcy nukleotydów biblioteka NuConf odtwarzała pozycje zasad z dokładnością porównywalną, a czasem lepszą niż standardowe biblioteki używane dla łańcuchów bocznych białek. Gdy program musiał wybierać kształty wyłącznie na podstawie energii, NuConf nadal przewyższała prostszą bibliotekę i konkurencyjne narzędzia dla kwasów nukleinowych, jednocześnie odzwierciedlając kluczowe kontakty parowania zasad i układania się w stosy zarówno w wolnych kwasach nukleinowych, jak i w kompleksach białko–kwas nukleinowy.
Co to oznacza dla przyszłego projektowania molekularnego
Dla osób niebędących specjalistami główny wniosek jest taki, że autorzy dali projektowaniu wspólny, nowy język zarówno dla białek, jak i dla kwasów nukleinowych. NuConf pozwala istniejącemu oprogramowaniu do projektowania białek wiarygodnie umieszczać i wybierać zasady DNA i RNA wzdłuż danego szkieletu oraz w miejscach kontaktu z białkami. Nie zastępuje to nowoczesnych metod AI, ale wypełnia ważną lukę, gdy danych treningowych jest niewiele lub gdy trzeba ocenić drobne, fizyczne oddziaływania. W dłuższej perspektywie tego typu narzędzie może pomóc badaczom projektować bardziej precyzyjne regulatory genów, przełączniki RNA oraz hybrydowe maszyny białkowo–kwasowe całkowicie in silico, zanim zostaną zbudowane w laboratorium.
Cytowanie: Makarova, M.O., Stiebritz, M.T., Basturk, D. et al. NuConf: a rotamer library for DNA and RNA and its implementation in the protein design software MUMBO. Sci Rep 16, 16281 (2026). https://doi.org/10.1038/s41598-026-52380-3
Słowa kluczowe: projektowanie kwasów nukleinowych, biblioteka rotamerów, interakcje białko–DNA, modelowanie RNA, obliczeniowa biologia strukturalna