Clear Sky Science · fr
NuConf : une bibliothèque de rotamères pour l'ADN et l'ARN et son intégration dans le logiciel de conception protéique MUMBO
Pourquoi remodeler l’ADN et l’ARN sur ordinateur compte
La conception de nouvelles protéines à l’aide d’outils informatiques a fait d’importants progrès ces dernières années, mais l’ADN et l’ARN sont restés largement en retrait. Les structures de ces molécules génétiques sont moins abondamment documentées, ce qui complique l’apprentissage par intelligence artificielle de leurs courbures, torsions et interactions avec les protéines. Cette étude présente NuConf, une nouvelle manière de représenter les formes des constituants de l’ADN et de l’ARN afin que les logiciels de conception protéique existants puissent également concevoir des acides nucléiques et leurs surfaces de contact avec des protéines. 
Comment les concepteurs traitent habituellement les parties latérales flexibles
Lorsque les scientifiques conçoivent des protéines sur ordinateur, ils n’explorent pas chaque mouvement possible de chaque atome des chaînes latérales. Ils utilisent plutôt des « bibliothèques de rotamères », des collections de formes communes de chaînes latérales extraites de milliers de structures connues. Des programmes comme MUMBO placent ces formes sur un backbone protéique fixe et utilisent des calculs d’énergie pour décider quelle combinaison s’ajuste le mieux. Jusqu’à présent, une bibliothèque pratique comparable pour les parties latérales des bases d’ADN et d’ARN faisait défaut, en particulier une qui permettrait au même programme de traiter protéines, ADN et ARN sur un pied d’égalité.
Cartographier les formes préférées de l’ADN et de l’ARN
Les auteurs ont commencé par examiner plus de 175 000 nucléotides issus de structures cristallographiques de haute qualité d’ADN, d’ARN et de leurs complexes avec des protéines. Pour chaque nucléotide, ils ont mesuré deux angles clés : l’un capturant la conformation de la pucker du sucre du backbone, et l’autre décrivant la rotation de la base attachée par rapport à ce sucre. Ils ont constaté que les sucres favorisent deux grandes familles de formes, l’une plus typique de l’ADN et l’autre de l’ARN, et que ces conformations du sucre sont étroitement liées à l’orientation de la base. Autrement dit, la posture du backbone et la direction de la base ne sont pas indépendantes ; elles évoluent de concert selon des modes caractéristique.
Transformer des motifs structuraux en une bibliothèque pratique
Pour convertir ces motifs en quelque chose qu’un programme de conception peut utiliser, l’équipe a appliqué des modèles statistiques qui décomposent des distributions d’angles compliquées en un petit nombre de pics distincts, chaque pic représentant une orientation fréquemment observée de la base. Pour chaque type de base en ADN et en ARN, et pour chaque grande conformation du sucre, ils ont défini trois à six orientations préférées, ainsi que leur fréquence d’occurrence. Cette collection, appelée NuConf, fonctionne comme un « catalogue de formes » de nucléosides lié à la posture locale du backbone. Ils ont aussi créé une version de secours plus simple avec moins de formes, qui sacrifie un peu de détail pour réduire le coût de calcul. 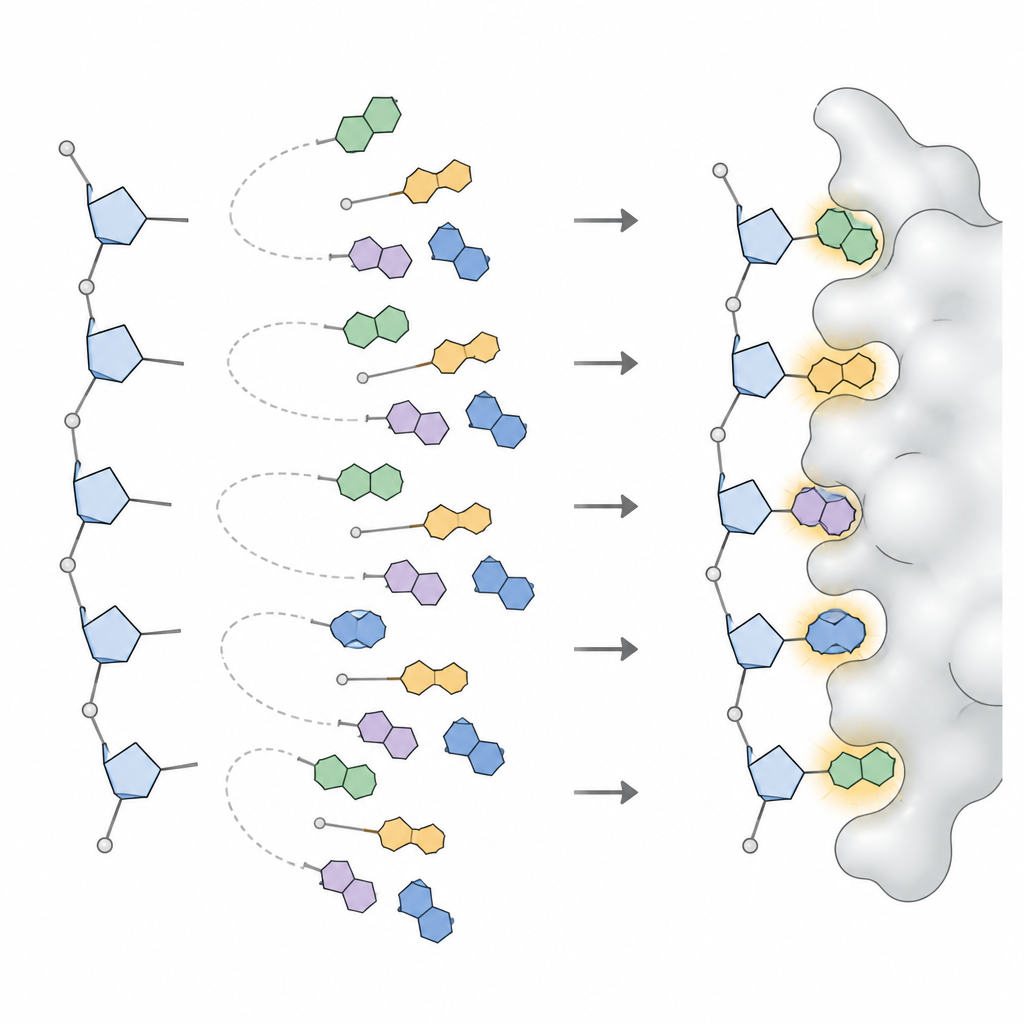
Vérifier si les nouvelles formes fonctionnent vraiment
Les chercheurs ont ensuite intégré ces formes dans MUMBO et posé deux questions : le programme, avec un backbone fixe, pouvait‑il reconstruire les positions de bases observées dans les structures réelles, et pouvait‑il choisir de bonnes formes et séquences en se basant uniquement sur des scores d’énergie, sans avoir la réponse ? Dans de larges jeux de test contenant des dizaines de milliers de nucléotides, la bibliothèque NuConf a reproduit les positions des bases avec une précision comparable, et parfois supérieure, à celle des bibliothèques standards utilisées pour les chaînes latérales protéiques. Lorsque le programme devait choisir les formes uniquement par énergie, NuConf a néanmoins surpassé une bibliothèque plus simple et des outils concurrents pour acides nucléiques, tout en capturant les contacts clés d’appariement et d’empilement des bases aussi bien dans les acides nucléiques libres que dans les complexes protéine–acide nucléique.
Ce que cela signifie pour la conception moléculaire future
Pour les non‑spécialistes, l’essentiel est que les auteurs ont fourni à la conception assistée par ordinateur un nouveau langage commun pour protéines et acides nucléiques. NuConf permet aux logiciels de conception protéique existants de placer et de sélectionner de manière fiable des bases d’ADN et d’ARN le long d’un backbone donné et sur les sites de contact protéiques. Cela ne remplace pas les méthodes modernes d’IA, mais comble une lacune importante lorsque les données d’entraînement sont rares ou lorsque des interactions physiques fines doivent être évaluées. À long terme, ce type d’outil pourrait aider les chercheurs à concevoir in silico des régulateurs de gènes plus précis, des commutateurs d’ARN et des machines hybrides protéine–acide nucléique avant leur construction en laboratoire.
Citation: Makarova, M.O., Stiebritz, M.T., Basturk, D. et al. NuConf: a rotamer library for DNA and RNA and its implementation in the protein design software MUMBO. Sci Rep 16, 16281 (2026). https://doi.org/10.1038/s41598-026-52380-3
Mots-clés: conception d’acides nucléiques, bibliothèque de rotamères, interactions protéine–ADN, modélisation de l’ARN, biologie structurale computationnelle