Clear Sky Science · sv
Jämförelse av djupet i shotgun-sekvensering avslöjar potentialen och begränsningarna hos ytlig metagenomik och analys på stamnivå
Varför studier av mikroliv behöver rätt mängd data
Microber som lever i och på oss påverkar vår hälsa, men de är för små och för mångskiftande för att räknas under mikroskop. Idag läser forskare ofta deras DNA för att se vilka mikrober som finns och vad de kan göra. Men mer DNA-data innebär högre kostnader. Denna studie ställer en enkel men viktig fråga: hur mycket DNA-sekvensering behövs egentligen för att få användbara svar om ett mikrobiellt community, och var börjar minskad insats leda till missvisande resultat?
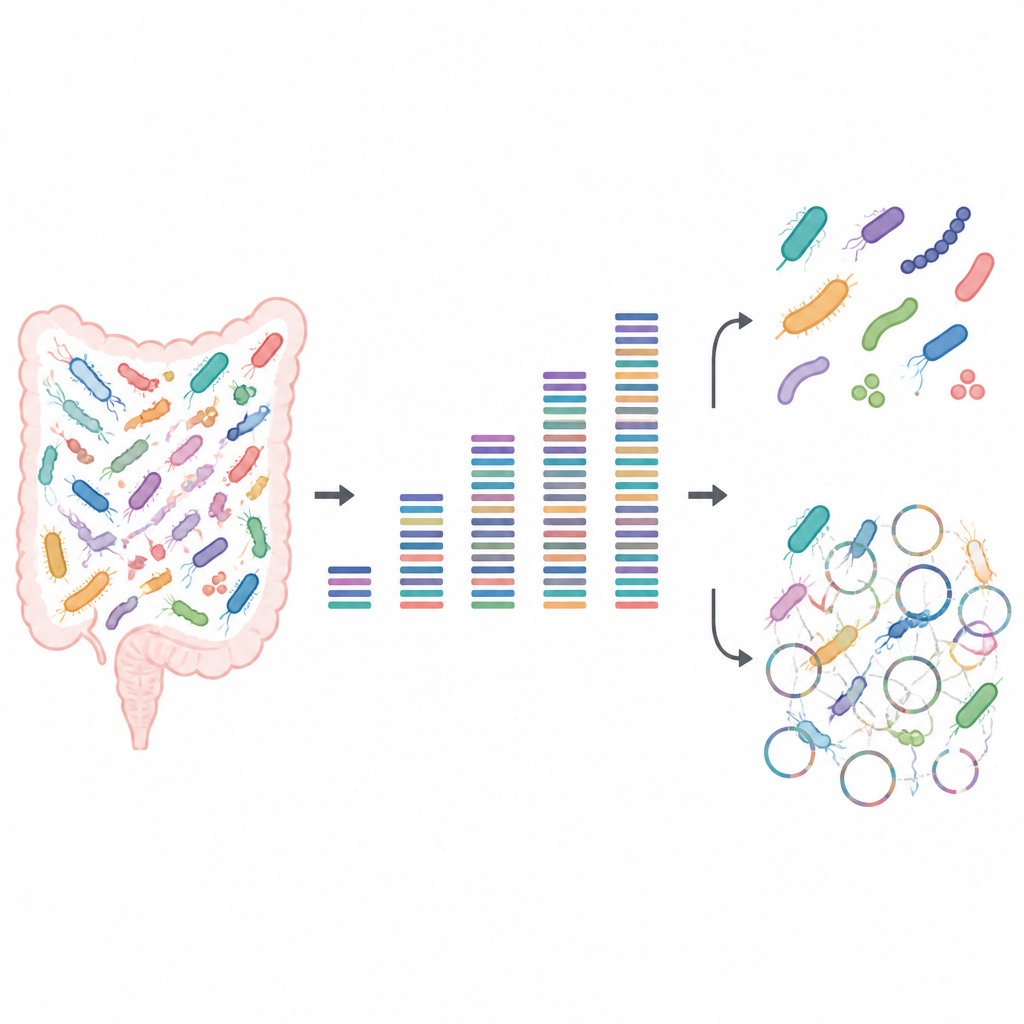
Test av styrkan hos små och stora datamängder
Forskarna byggde konstgjorda mikrobiella communityn av kända tarmbakterier odlade i labb. Eftersom de visste exakt vilka stammar som fanns och i vilka mängder fungerade dessa ”mock”-prover som ett testmönster för en tv-skärm, och avslöjade var bilden från DNA-sekvenseringen var skarp eller suddig. De sekvenserade varje community i djup som sträckte sig från mycket små datamängder till mycket stora, och analyserade sedan vilka bakterier de kunde upptäcka, om nära besläktade stammar kunde särskiljas, och hur mycket av varje stams proteinproducerande potential de kunde återfinna.
Vad som fungerar bra med lite sekvensering
För grundläggande frågor som ”vilka arter finns här” och ”hur vanliga är de” fann teamet att förvånansvärt lite data krävdes när bra referensgenom fanns tillgängliga. Även vid låg sekvenseringsdjup lämnade varje stam ett upptäckbart spår, och relativa abundansmönster förblev stabila när mer data adderades. Ungefär en halv gigabase DNA per prov räckte för att pålitligt kartlägga communityns medlemskap. Det gör låg-djup eller ”ytlig” sekvensering attraktivt för stora studier som vill jämföra övergripande mikrobiommönster över många personer eller tillstånd utan att spendera en förmögenhet.
Var ytlig sekvensering brister
Problem uppstod när fokus flyttades från arter till individuella stammar och deras detaljerade funktioner. Att återskapa hela genom från grunden, en process som kallas metagenomassembly, krävde mycket djupare sekvensering och gick ändå ofta fel. Datorprogram grupperade DNA-fragment till utkast till genom som såg ut att vara hög kvalitet enligt standardkontroller, men många av dessa var i själva verket lapptäcken sammansatta av flera olika stammar. Även vid mycket höga sekvenseringsdjup var en betydande andel av de assemblerade genomen kimeriska, och vissa verkliga stammar missades helt. Ytlig sekvensering hade också svårt att fånga hela uppsättningen proteiner: medan några få gigabaser räckte för att skissa breda metaboliska vägar, krävdes betydligt djupare sekvensering för att täcka de flesta enskilda proteiner, särskilt i mer komplexa communityn.
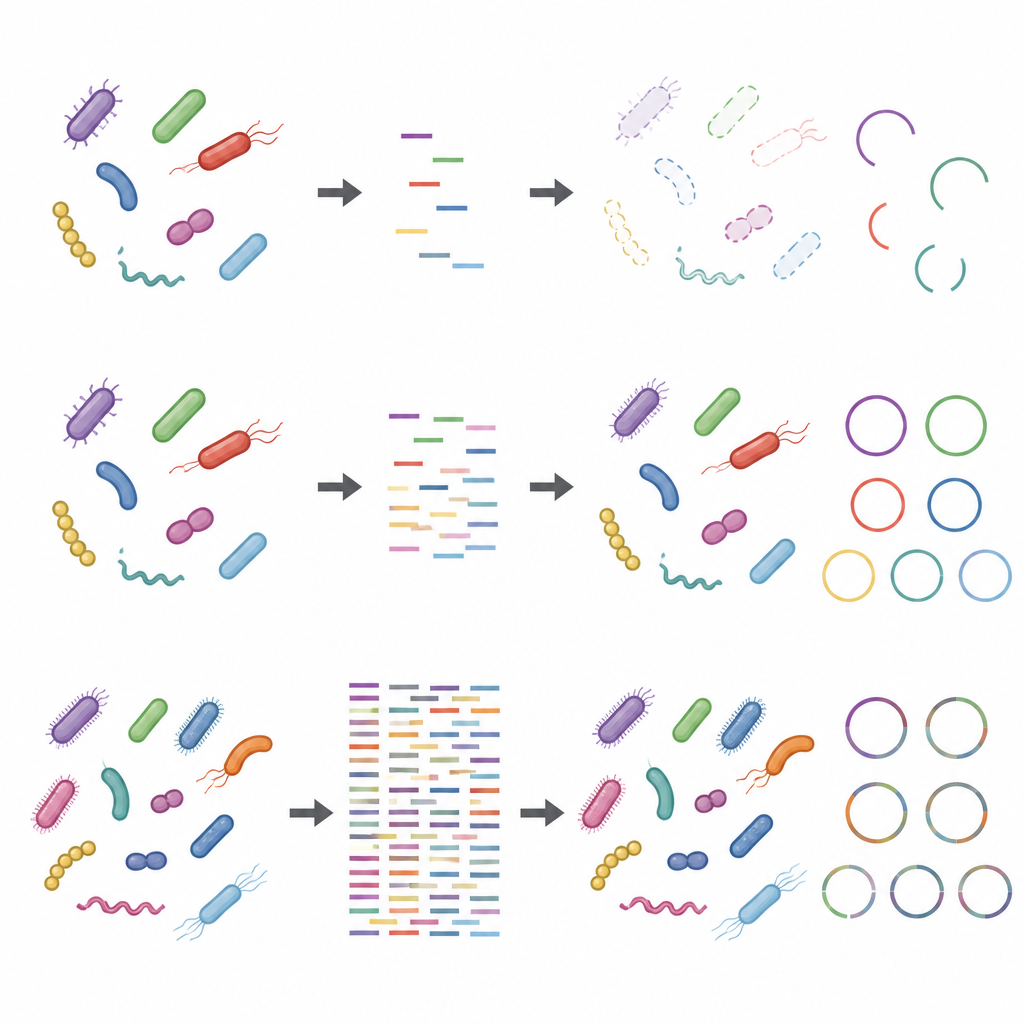
Effekter av laboratorieval och främmande DNA
Studien visade också att steg som utförs innan DNA:t når sekvenseraren kan förvränga resultaten, särskilt vid lågt sekvenseringsdjup. Att använda mer startmaterial och färre amplifieringsrundor gjorde taxonomiska och funktionella profiler mer robusta. I kontrast förvrängde protokoll med lite ingångs-DNA och många amplifieringscykler den relativa abundansen av vissa stammar. Att tillsätta värd-DNA, vilket efterliknar vad som händer i prover rika på mänskligt eller animaliskt material, minskade dessutom den uppenbara täckningen av mikrobiella genom och deras proteiner. Dessa problem blev mindre allvarliga vid högre sekvenseringsdjup, men försvann inte helt.
Praktisk vägledning för framtida mikrobiomstudier
Sammanfattningsvis erbjuder arbetet en verklighetskontroll för vad ytlig DNA-sekvensering kan och inte kan leverera. För studier som främst behöver en bred förteckning över vilka mikrober som finns i en välstuderad miljö, såsom människans tarm, kan måttliga sekvenseringsdjup fungera väl, förutsatt att bra referensgenom finns och att laboratorieprotokoll väljs omsorgsfullt. För detaljerade frågor om en communitys funktioner eller finskaliga skillnader mellan stammar räcker inte ytlig sekvensering. Även mycket djup sekvensering kan inte helt lösa problemet med sammanblandade assemblerade genom, så resultat baserade på sådana utkast bör behandlas med försiktighet. Kort sagt: mängden DNA-data och analysmetoderna bör anpassas till den vetenskapliga frågan, med en klar förståelse för var bilden förblir suddig.
Citering: Treichel, N.S., Pauvert, C., Séneca, J. et al. Benchmarking of shotgun sequencing depth reveals the potential and limitations of shallow metagenomics and strain-level analysis. Nat Microbiol 11, 1233–1244 (2026). https://doi.org/10.1038/s41564-026-02334-2
Nyckelord: mikrobiomsekvensering, ytlig metagenomik, sekvenseringsdjup, metagenom-assemblerade genom, analys på stamnivå