Clear Sky Science · nl
Benchmarking van shotgun-sequenceringsdiepte onthult de mogelijkheden en beperkingen van vlakke metagenomica en analyse op soortniveau
Waarom het bekijken van minuscule levensvormen de juiste hoeveelheid data vereist
Microben die in en op ons leven beïnvloeden onze gezondheid, maar ze zijn veel te klein en divers om onder een microscoop te tellen. Tegenwoordig lezen wetenschappers vaak hun DNA om te bepalen welke microben aanwezig zijn en wat ze kunnen doen. Meer DNA-data betekent echter hogere kosten. Deze studie stelt een eenvoudige maar belangrijke vraag: hoeveel DNA-sequencing is werkelijk nodig om bruikbare antwoorden over een microbieel ecosysteem te krijgen, en vanaf welk punt begint bezuinigen ons op het verkeerde been te zetten?
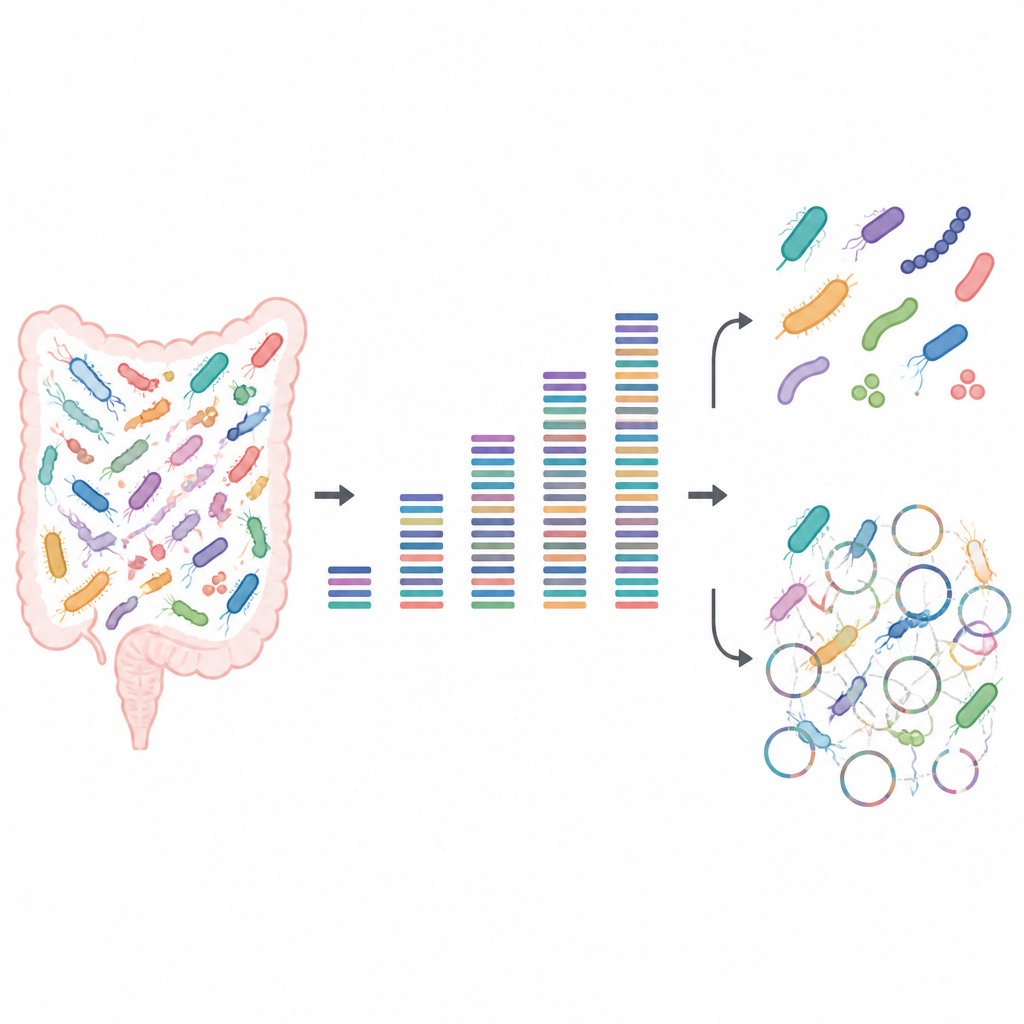
Het vermogen van kleine en grote datasets testen
De onderzoekers stelden kunstmatige microbieele gemeenschappen samen uit bekende darmbacteriën die in het laboratorium gekweekt waren. Omdat ze precies wisten welke stammen aanwezig waren en in welke hoeveelheden, fungeerden deze "mock"-monsters als een testpatroon voor een televisiescherm, en lieten ze zien waar het beeld van DNA-sequencing scherp of onscherp was. Ze sequenceden elke gemeenschap op dieptes variërend van zeer weinig tot zeer veel data en analyseerden vervolgens welke bacteriën ze konden detecteren, of nauw verwante stammen uit elkaar gehouden konden worden en hoeveel van het eiwitproducerende potentieel van elke stam ze konden terugvinden.
Wat goed werkt bij weinig sequencing
Voor basisvragen zoals "welke soorten zijn hier" en "hoe algemeen is elke soort" bleek verrassend weinig data nodig wanneer goede referentiegenomen beschikbaar waren. Zelfs bij lage sequencingdiepte liet elke stam een detecteerbare trace achter en bleven patronen van relatieve aantallen stabiel naarmate er meer data bijkwamen. Ongeveer een halve gigabase DNA per monster bleek voldoende om de samenstelling van de gemeenschap betrouwbaar in kaart te brengen. Dit maakt ondiepe, ofwel "shallow", sequencing aantrekkelijk voor grootschalige studies die algemene microbioompatronen willen vergelijken over veel personen of condities zonder enorme kosten.
Waar ondiepe benaderingen tekortschieten
Problemen deden zich voor zodra de focus verschoof van soorten naar individuele stammen en hun gedetailleerde functies. Het reconstrueren van volledige genomen vanaf nul, een proces dat bekendstaat als metagenoom-assemblage, vergde veel diepere sequencing en ging nog vaak mis. Computerprogramma's groeperden DNA-fragmenten tot conceptuele genomen die er volgens gangbare controles als hoge kwaliteit uitzagen, maar veel van deze genomen waren in feite lappendekens samengesteld uit meerdere verschillende stammen. Zelfs bij zeer diepe sequencing bleven een aanzienlijk aantal van deze geassembleerde genomen chimerisch en sommige echte stammen werden helemaal gemist. Ondiepe sequencing had ook moeite om de volledige set aanwezige eiwitten vast te leggen: terwijl een paar gigabases genoeg waren om brede metabole routes te schetsen, was veel diepere sequencing vereist om de meeste individuele eiwitten te dekken, vooral in complexere gemeenschappen.
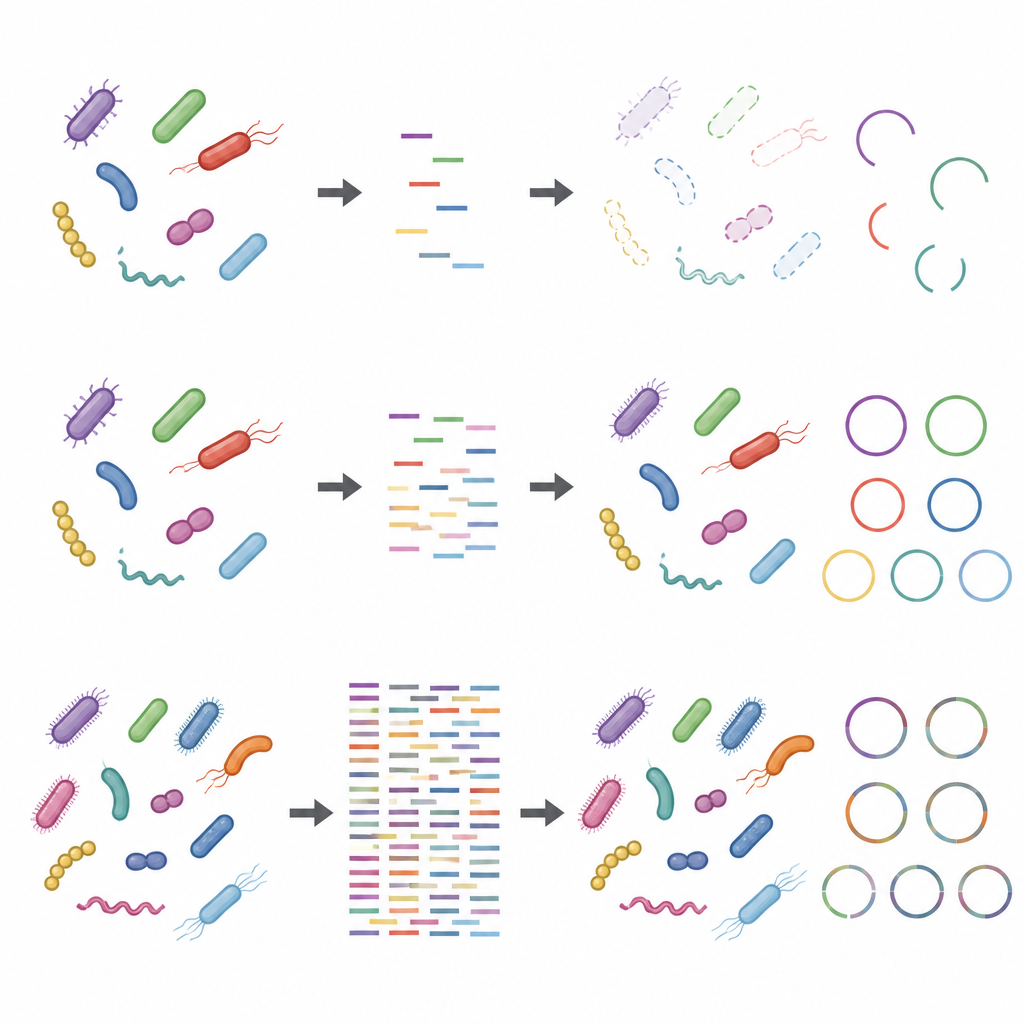
Effecten van laboratoriumkeuzes en vreemde DNA
De studie toonde ook aan dat stappen die worden genomen voordat het DNA de sequencer bereikt de resultaten kunnen vertekenen, vooral wanneer de sequencingdiepte laag is. Het gebruik van meer begindna en minder amplificatiecycli maakte taxonomische en functionele profielen robuuster. Daarentegen verstoorden protocollen met weinig input-DNA en veel amplificatiecycli de relatieve aantallen van sommige stammen. Het toevoegen van gastheer-DNA, wat nabootst wat er gebeurt bij monsters rijk aan menselijk of dierlijk materiaal, verminderde de schijnbare dekking van microbiele genomen en hun eiwitten verder. Deze problemen werden minder ernstig bij hogere sequencingdieptes, maar verdwenen niet volledig.
Praktische richtlijnen voor toekomstige microbioomstudies
Samengevat biedt dit werk een realiteitscheck over wat ondiepe DNA-sequencing wel en niet kan leveren. Voor studies die voornamelijk een brede inventarisatie nodig hebben van welke microben aanwezig zijn in een goed bestudeerde omgeving zoals de menselijke darm, kunnen bescheiden sequencingdieptes goed genoeg zijn, mits goede referentiegenomen bestaan en laboratoriumprotocollen zorgvuldig zijn gekozen. Voor gedetailleerde vragen over de functies van een gemeenschap of de fijnmazige verschillen tussen stammen is ondiepe sequencing echter onvoldoende. Zelfs zeer diepe sequencing kan het probleem van vermengde geassembleerde genomen niet volledig oplossen, dus resultaten op basis van dergelijke conceptgenomen moeten met voorzichtigheid worden geïnterpreteerd. Kortom: de hoeveelheid DNA-data en de analysemethoden moeten worden afgestemd op de wetenschappelijke vraag, met een duidelijk begrip van waar het beeld wazig blijft.
Bronvermelding: Treichel, N.S., Pauvert, C., Séneca, J. et al. Benchmarking of shotgun sequencing depth reveals the potential and limitations of shallow metagenomics and strain-level analysis. Nat Microbiol 11, 1233–1244 (2026). https://doi.org/10.1038/s41564-026-02334-2
Trefwoorden: microbioom-sequencing, vlakke metagenomica, sequenceringsdiepte, metagenoom-geassembleerde genomen, analyse op strain-niveau