Clear Sky Science · en
Benchmarking of shotgun sequencing depth reveals the potential and limitations of shallow metagenomics and strain-level analysis
Why looking at tiny life needs the right data size
Microbes living in and on us shape our health, yet they are far too small and diverse to count under a microscope. Today, scientists often read their DNA to see which microbes are present and what they can do. But more DNA data means higher costs. This study asks a simple but important question: how much DNA sequencing is really needed to get useful answers about a microbial community, and where does cutting corners start to mislead us?
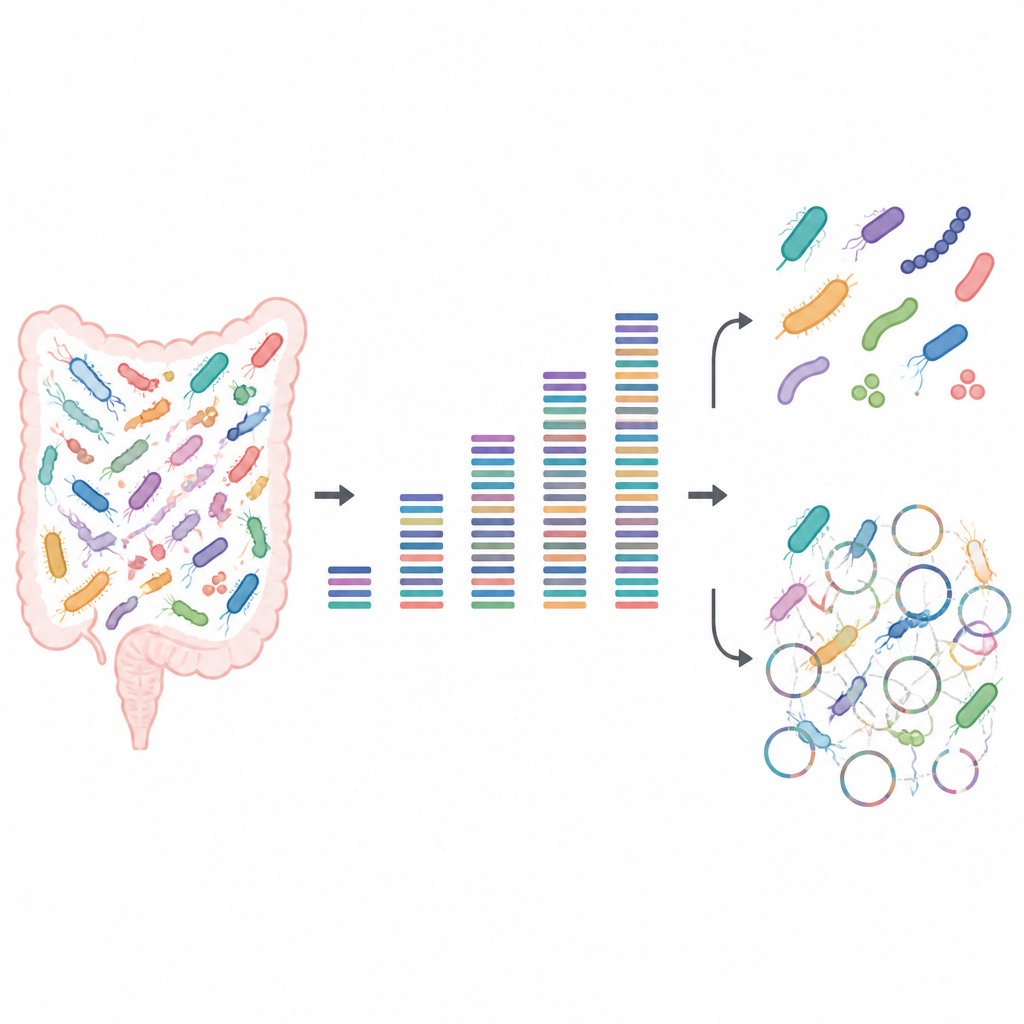
Testing the power of small and big data
The researchers built artificial microbial communities from known gut bacteria grown in the lab. Because they knew exactly which strains were present and in what amounts, these "mock" samples acted like a test pattern for a television screen, revealing where the picture from DNA sequencing was sharp or blurry. They sequenced each community at depths ranging from a very small amount of data to very large, and then analysed which bacteria they could detect, whether closely related strains could be told apart, and how much of each strain’s protein-making potential they could recover.
What works well with little sequencing
For basic questions such as "which species are here" and "how common is each one," the team found that surprisingly little data was needed when good reference genomes were available. Even at low sequencing depth, every strain left a detectable trace, and relative abundance patterns stayed stable as more data were added. Around half a gigabase of DNA per sample was enough to profile community membership reliably. This makes low-depth, or "shallow," sequencing attractive for large studies that want to compare overall microbiome patterns across many people or conditions without spending a fortune.
Where shallow approaches fall short
Problems appeared once the focus shifted from species to individual strains and their detailed functions. Rebuilding whole genomes from scratch, a process known as metagenome assembly, needed much deeper sequencing and still often went wrong. Computer programs grouped DNA fragments into draft genomes that looked high quality by standard checklists, yet many of these were actually patchworks made from several different strains. Even at very high sequencing depths, a notable share of these assembled genomes were chimeric, and some true strains were missed altogether. Shallow sequencing also struggled to capture the full set of proteins present: while a few gigabases were enough to outline broad metabolic pathways, far deeper sequencing was required to cover most individual proteins, especially in more complex communities.
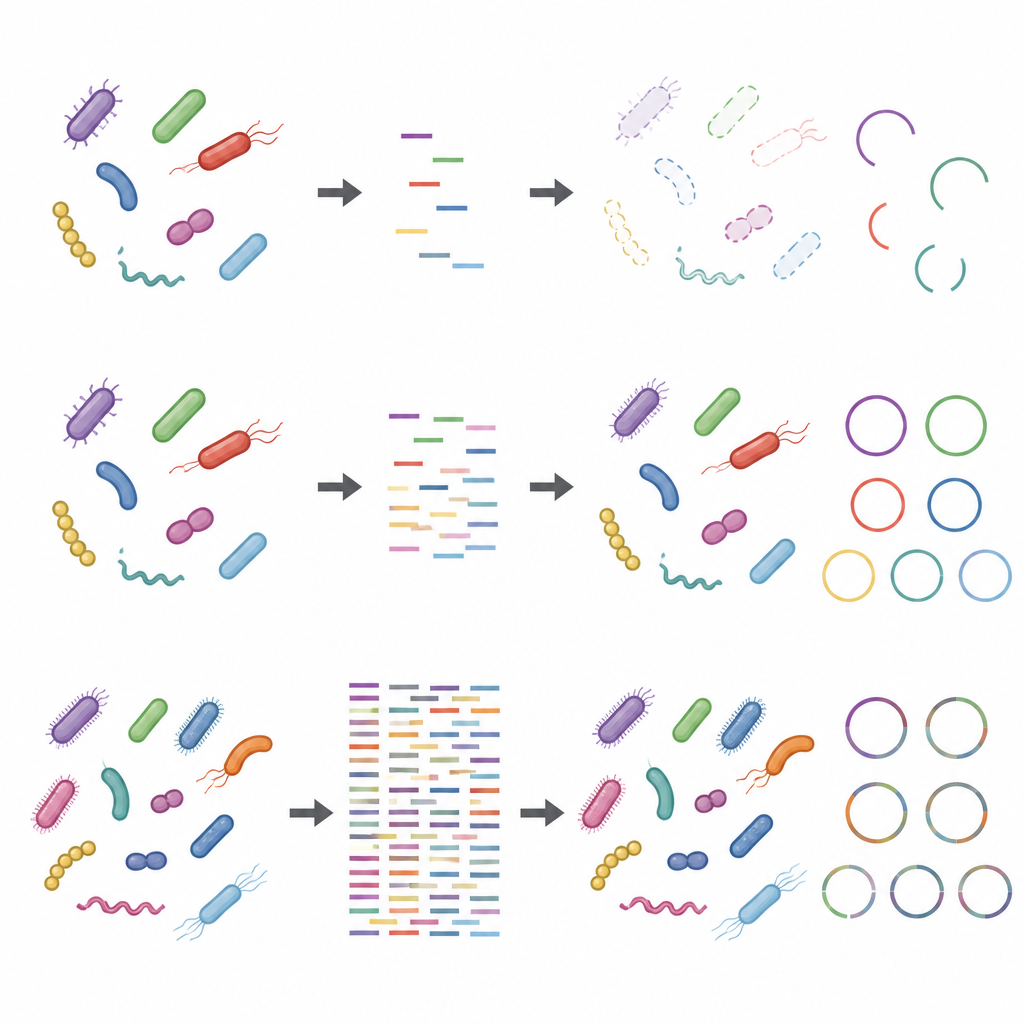
Effects of lab choices and stray DNA
The study also showed that steps taken before the DNA ever reaches the sequencer can skew results, particularly when sequencing depth is low. Using more starting DNA and fewer rounds of amplification made taxonomic and functional profiles more robust. In contrast, protocols with little input DNA and many amplification cycles distorted the relative abundance of some strains. Adding host DNA, mimicking what happens with samples rich in human or animal material, further reduced the apparent coverage of microbial genomes and their proteins. These issues became less serious at higher sequencing depths, but did not disappear entirely.
Practical guidance for future microbiome studies
Overall, the work offers a reality check on what shallow DNA sequencing can and cannot deliver. For studies that mainly need a broad census of which microbes are present in a well-studied environment such as the human gut, modest sequencing depths can work well, provided good reference genomes exist and lab protocols are carefully chosen. However, for detailed questions about the functions of a community or the fine-scale differences between strains, shallow sequencing is not enough. Even very deep sequencing cannot fully fix the problem of mixed-up assembled genomes, so results based on such drafts must be treated with caution. In short, the amount of DNA data and the analysis methods should be matched to the scientific question, with a clear understanding of where the picture remains fuzzy.
Citation: Treichel, N.S., Pauvert, C., Séneca, J. et al. Benchmarking of shotgun sequencing depth reveals the potential and limitations of shallow metagenomics and strain-level analysis. Nat Microbiol 11, 1233–1244 (2026). https://doi.org/10.1038/s41564-026-02334-2
Keywords: microbiome sequencing, shallow metagenomics, sequencing depth, metagenome-assembled genomes, strain-level analysis