Clear Sky Science · pl
Porównanie głębokości sekwencjonowania shotgun ujawnia możliwości i ograniczenia płytkiej metagenomiki oraz analizy na poziomie szczepów
Dlaczego badanie mikroskopijnego życia wymaga odpowiedniej ilości danych
Mikroby żyjące w i na nas kształtują nasze zdrowie, a jednocześnie są zbyt małe i zróżnicowane, by je policzyć pod mikroskopem. Dziś naukowcy często odczytują ich DNA, by ustalić, które mikroby występują i jakie mają możliwości. Więcej danych DNA oznacza jednak wyższe koszty. To badanie stawia proste, lecz istotne pytanie: ile sekwencjonowania DNA naprawdę potrzeba, by uzyskać użyteczne odpowiedzi o społeczności mikroorganizmów, i gdzie oszczędzanie zaczyna wprowadzać w błąd?
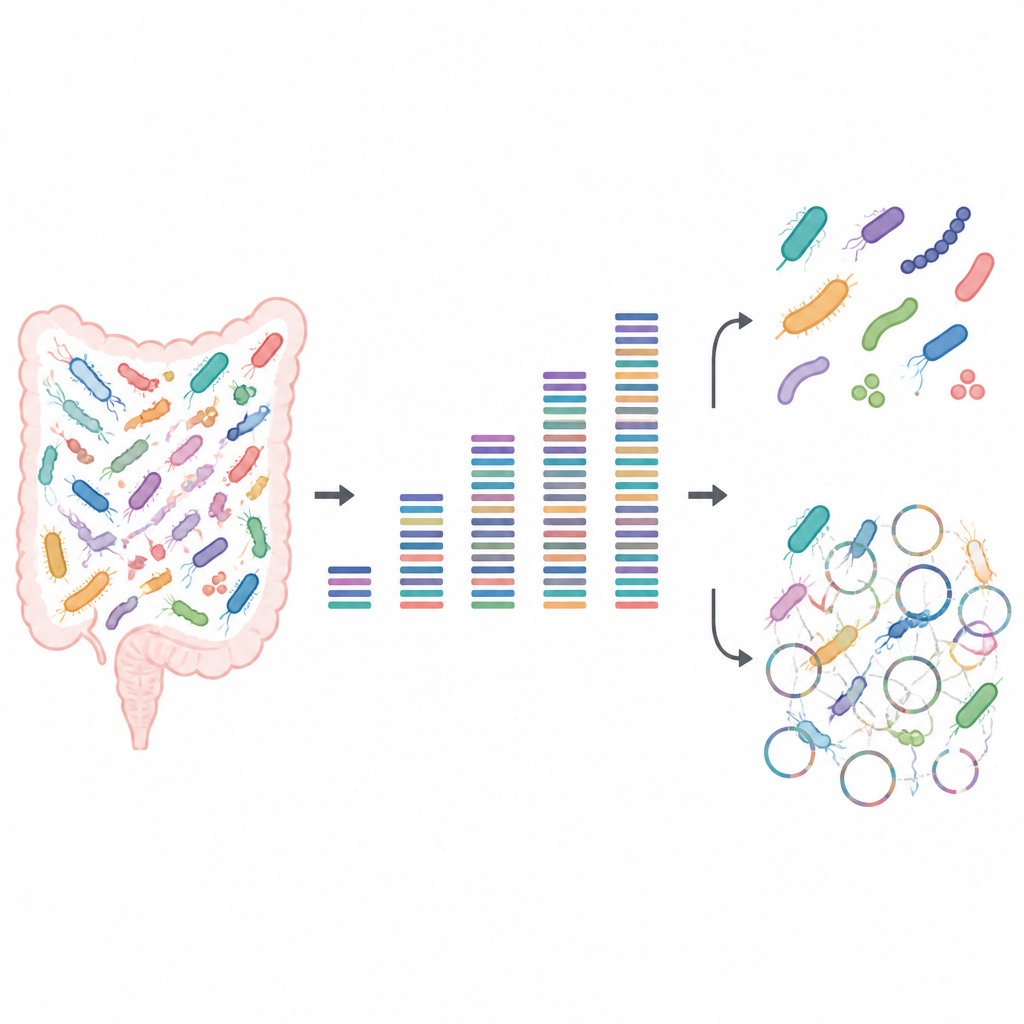
Testowanie siły małych i dużych danych
Naukowcy zbudowali sztuczne społeczności mikrobiologiczne z poznanych bakterii jelitowych hodowanych w laboratorium. Ponieważ dokładnie wiedzieli, które szczepy są obecne i w jakich ilościach, te „wzorcowe” próbki działały jak testowy wzór dla ekranu telewizora, odsłaniając, gdzie obraz uzyskany dzięki sekwencjonowaniu DNA był ostry, a gdzie rozmyty. Sekwencjonowali każdą społeczność przy różnych głębokościach — od bardzo małej ilości danych po bardzo dużą — a następnie analizowali, które bakterie udało się wykryć, czy da się rozróżnić blisko spokrewnione szczepy i jak dużo potencjału do produkcji białek każdego szczepu udało się odzyskać.
Co działa dobrze przy niewielkim sekwencjonowaniu
Dla podstawowych pytań, takich jak „jakie gatunki tu są” i „jak częsty jest każdy z nich”, zespół odkrył, że zaskakująco niewiele danych wystarcza, gdy dostępne są dobre genomy referencyjne. Nawet przy niskiej głębokości sekwencjonowania każdy szczep pozostawiał wykrywalny ślad, a wzorce względnej obfitości pozostawały stabilne wraz z dodawaniem danych. Około połowy gigabajta DNA na próbkę było wystarczające do wiarygodnego określenia członkostwa w społeczności. To sprawia, że płytkie sekwencjonowanie jest atrakcyjne dla dużych badań, które chcą porównywać ogólne wzorce mikrobiomu wśród wielu osób lub warunków bez ponoszenia ogromnych kosztów.
Gdzie podejścia płytkie zawodzą
Problemy pojawiały się, gdy uwaga przenosiła się ze skali gatunku na poszczególne szczepy i ich szczegółowe funkcje. Odtworzenie całych genomów od podstaw, proces znany jako składanie metagenomu, wymagało znacznie głębszego sekwencjonowania i nadal często zawodziło. Programy komputerowe grupowały fragmenty DNA w szkicowe genomy, które wyglądały na wysokiej jakości według standardowych list kontrolnych, jednak wiele z nich było w rzeczywistości patchworkami złożonymi z kilku różnych szczepów. Nawet przy bardzo wysokich głębokościach sekwencjonowania znacząca część złożonych genomów była chimeryczna, a niektóre prawdziwe szczepy zostały całkowicie pominięte. Płytkie sekwencjonowanie miało także trudności z uchwyceniem pełnego zestawu białek: podczas gdy kilka gigabajtów było wystarczających, by zarysować szerokie ścieżki metaboliczne, znacznie głębsze sekwencjonowanie było potrzebne, by pokryć większość pojedynczych białek, szczególnie w bardziej złożonych społecznościach.
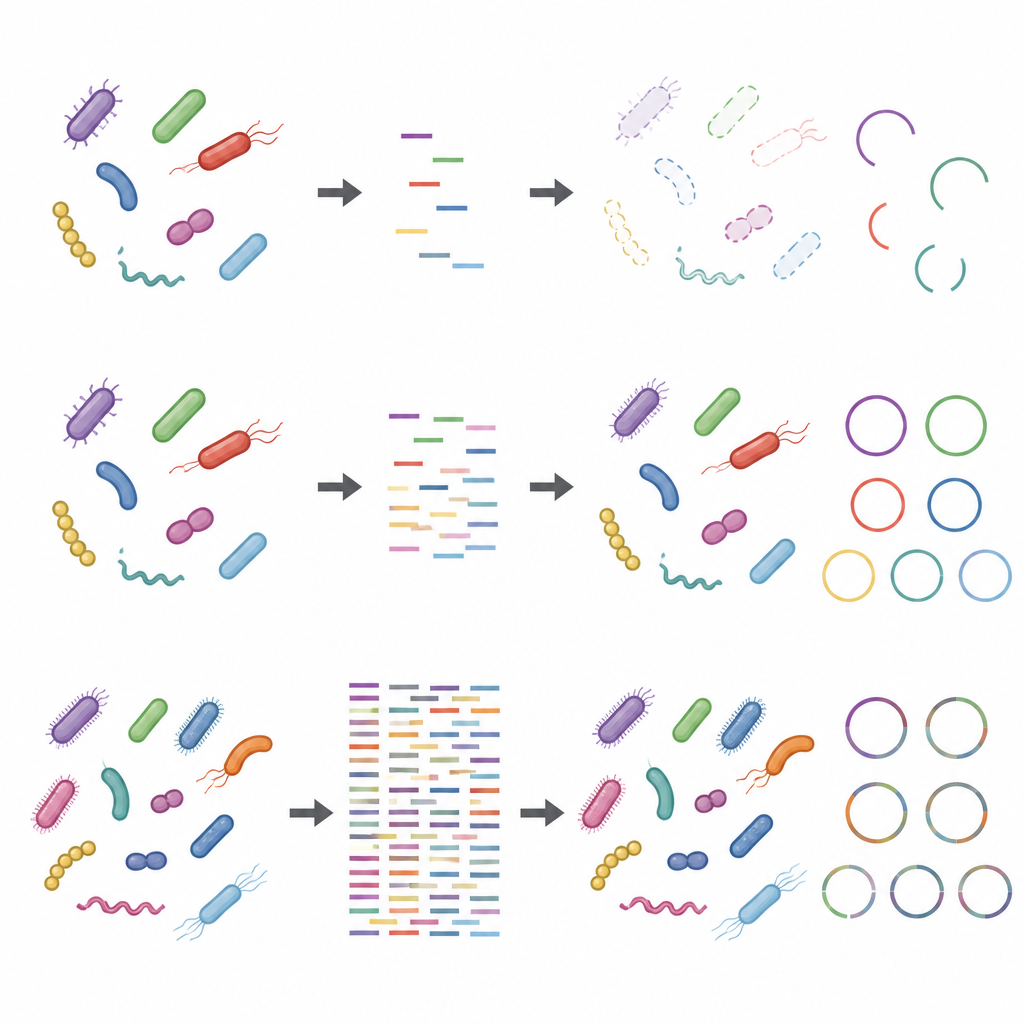
Wpływ wyborów laboratoryjnych i obcego DNA
Badanie pokazało również, że kroki podejmowane zanim DNA trafi do sekwencera mogą zniekształcać wyniki, szczególnie przy niskiej głębokości sekwencjonowania. Użycie większej ilości DNA wyjściowego i mniejszej liczby cykli amplifikacji uczyniło profile taksonomiczne i funkcjonalne bardziej odpornymi. W przeciwieństwie do tego protokoły z małą ilością materiału wyjściowego i wieloma cyklami amplifikacji zniekształcały względne obfitości niektórych szczepów. Dodanie DNA gospodarza, naśladujące próbki bogate w materiał ludzki lub zwierzęcy, dodatkowo zmniejszało pozorną pokrycie genomów mikroorganizmów i ich białek. Problemy te stawały się mniej dotkliwe przy wyższych głębokościach sekwencjonowania, ale nie znikały całkowicie.
Praktyczne wskazówki dla przyszłych badań mikrobiomu
Podsumowując, praca daje realistyczne spojrzenie na to, co płytkie sekwencjonowanie DNA może, a czego nie może dostarczyć. Dla badań, które głównie potrzebują szerokiego spisu, jakie mikroby występują w dobrze poznanym środowisku, takim jak ludzki jelit, umiarkowane głębokości sekwencjonowania mogą sprawdzać się dobrze, pod warunkiem istnienia dobrych genomów referencyjnych i starannego doboru procedur laboratoryjnych. Jednak do szczegółowych pytań o funkcje społeczności lub drobne różnice między szczepami płytkie sekwencjonowanie nie wystarczy. Nawet bardzo głębokie sekwencjonowanie nie rozwiązuje całkowicie problemu pomieszanych złożonych genomów, więc wyniki oparte na takich szkicach należy traktować ostrożnie. Krótko mówiąc, ilość danych DNA i zastosowane metody analizy powinny być dopasowane do pytania naukowego, z jasnym rozeznaniem, gdzie obraz pozostaje nieostry.
Cytowanie: Treichel, N.S., Pauvert, C., Séneca, J. et al. Benchmarking of shotgun sequencing depth reveals the potential and limitations of shallow metagenomics and strain-level analysis. Nat Microbiol 11, 1233–1244 (2026). https://doi.org/10.1038/s41564-026-02334-2
Słowa kluczowe: sekwencjonowanie mikrobiomu, płytka metagenomika, głębokość sekwencjonowania, genomy złożone z metagenomów, analiza na poziomie szczepu