Clear Sky Science · ru
Бенчмаркинг глубины шотган-секвенирования выявляет потенциал и ограничения неглубокой метагеномики и анализа на уровне штаммов
Почему для изучения крошечной жизни нужен правильный объём данных
Микробы, живущие в нас и на нас, формируют наше здоровье, но они слишком малы и разнообразны, чтобы пересчитать их под микроскопом. Сегодня учёные часто считывают их ДНК, чтобы понять, какие микроорганизмы присутствуют и что они могут делать. Но больше данных ДНК означает и большие затраты. В этом исследовании ставится простой, но важный вопрос: сколько секвенирования ДНК действительно нужно, чтобы получить полезные ответы о микробном сообществе, и где экономия начинает вводить в заблуждение?
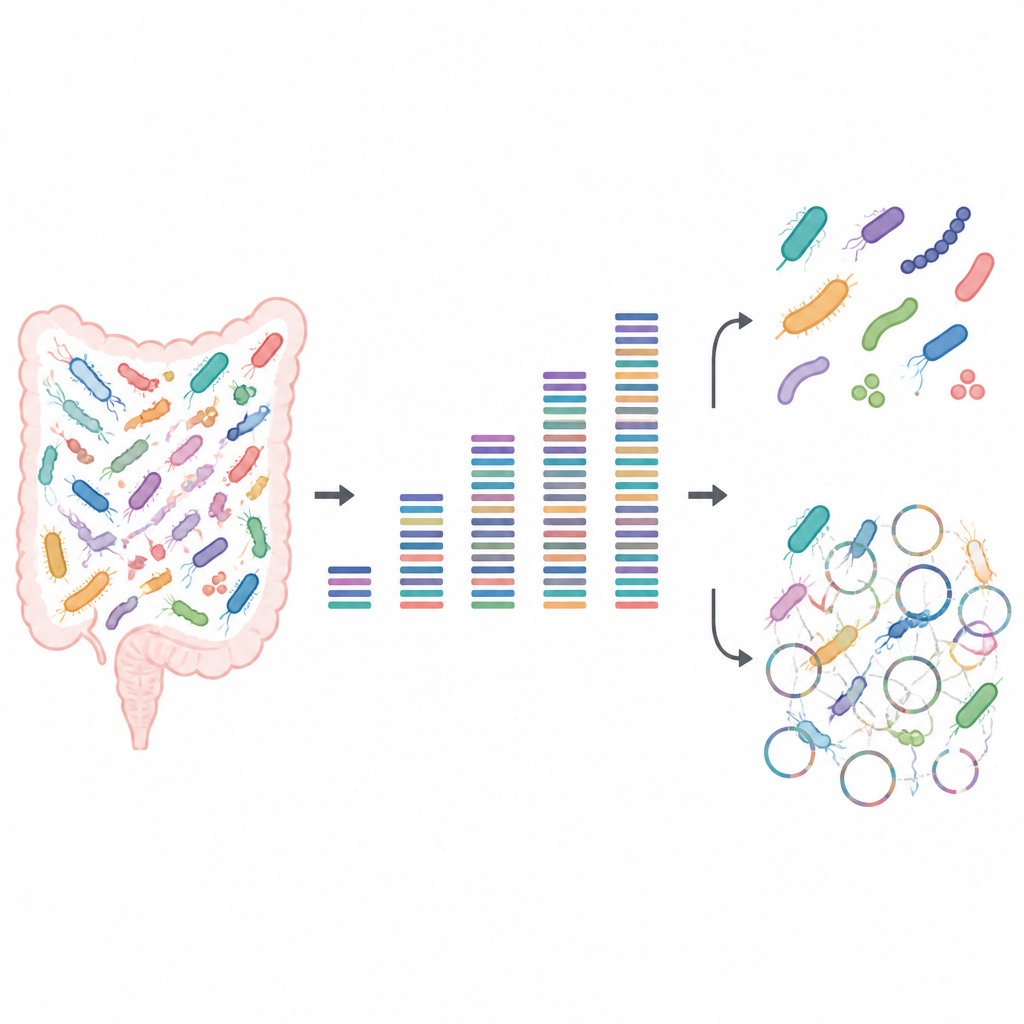
Тестирование силы маленьких и больших данных
Исследователи создали искусственные микробные сообщества из известных кишечных бактерий, выращенных в лаборатории. Поскольку они точно знали, какие штаммы присутствуют и в каких количествах, эти «мок‑» образцы выступали в роли тестовой таблицы для телевизора, показывая, где картина, полученная из секвенирования ДНК, резкая, а где — размытая. Они секвенировали каждое сообщество с глубинами, варьировавшими от очень малых объёмов данных до очень больших, а затем анализировали, какие бактерии можно обнаружить, можно ли различить тесно родственные штаммы и сколько от каждого штамма их белкового потенциала можно восстановить.
Что хорошо работает при небольшом объёме секвенирования
Для базовых вопросов, таких как «какие виды здесь присутствуют» и «насколько распространён каждый из них», команда обнаружила, что при наличии хороших референсных геномов требуется удивительно мало данных. Даже при низкой глубине секвенирования каждый штамм оставлял обнаружимый след, а относительные паттерны распространённости оставались устойчивыми с добавлением данных. Приблизительно полгигабазы ДНК на образец было достаточно для надёжного профилирования состава сообщества. Это делает неглубокое, или «shallow», секвенирование привлекательным для крупных исследований, которые хотят сравнивать общие паттерны микробиома среди большого числа людей или условий без больших затрат.
Где неглубокие подходы терпят неудачу
Проблемы возникали, как только фокус смещался от видов к отдельным штаммам и их детальным функциям. Восстановление целых геномов «с нуля», процесс, известный как метагеномная сборка, требовало гораздо более глубокой глубины секвенирования и при этом часто давало неправильные результаты. Программы группировали фрагменты ДНК в черновые геномы, которые по стандартным проверкам выглядели высокого качества, но многие из них на самом деле были мозаикой, составленной из нескольких разных штаммов. Даже при очень большой глубине секвенирования заметная доля таких собранных геномов оказывалась химерной, а некоторые истинные штаммы вовсе не выявлялись. Неглубокое секвенирование также плохо охватывало полный набор присутствующих белков: несколько гигабайт данных было достаточно, чтобы описать широкие метаболические пути, но для покрытия большинства отдельных белков, особенно в более сложных сообществах, требовалось существенно больше данных.
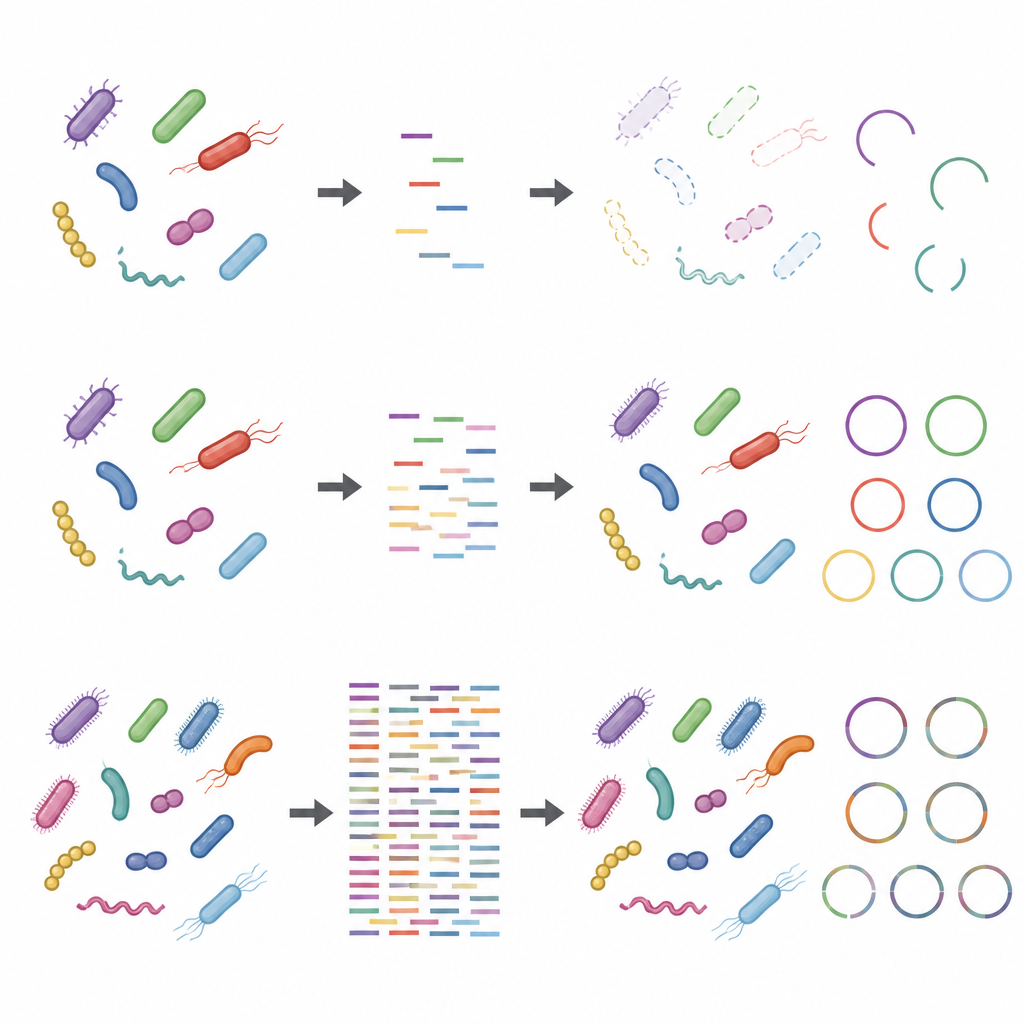
Влияние лабораторных решений и посторонней ДНК
Исследование также показало, что шаги до попадания ДНК в секвенсор могут искажать результаты, особенно при низкой глубине секвенирования. Использование большего количества исходной ДНК и меньшего числа циклов амплификации делало таксономические и функциональные профили более устойчивыми. Напротив, протоколы с малым входным количеством ДНК и множественными циклами амплификации искажали относительную распространённость некоторых штаммов. Добавление ДНК хозяина, имитирующее образцы, богатые человеческим или животным материалом, дополнительно снижало кажущуюся покрываемость микробных геномов и их белков. Эти проблемы становились менее серьёзными при более высокой глубине секвенирования, но полностью не исчезали.
Практические рекомендации для будущих исследований микробиома
В целом работа даёт приземлённую оценку того, что может и чего не может дать неглубокое секвенирование ДНК. Для исследований, которым в основном нужен общий учёт присутствующих микробов в хорошо изученной среде, например в человеческом кишечнике, умеренные глубины секвенирования могут работать хорошо при условии наличия хороших референсных геномов и корректно выбранных лабораторных протоколов. Однако для детальных вопросов о функциях сообщества или о тонких различиях между штаммами неглубокого секвенирования недостаточно. Даже очень глубокое секвенирование не в состоянии полностью устранить проблему смешанных собранных геномов, поэтому результаты, основанные на таких черновиках, следует интерпретировать с осторожностью. Короче говоря, объём данных ДНК и методы анализа должны соответствовать научному вопросу с ясным пониманием того, где изображение остаётся нечетким.
Цитирование: Treichel, N.S., Pauvert, C., Séneca, J. et al. Benchmarking of shotgun sequencing depth reveals the potential and limitations of shallow metagenomics and strain-level analysis. Nat Microbiol 11, 1233–1244 (2026). https://doi.org/10.1038/s41564-026-02334-2
Ключевые слова: секвенирование микробиома, неглубокая метагеномика, глубина секвенирования, собранные метагеномные геномы, анализ на уровне штаммов