Clear Sky Science · de
Benchmarking der Shotgun-Sequenzierungstiefe zeigt Potenziale und Grenzen von flacher Metagenomik und Analysen auf Stammebene
Warum das Betrachten winzigen Lebens die richtige Datenmenge braucht
Mikroben, die in und auf uns leben, prägen unsere Gesundheit, sind aber viel zu klein und zu vielfältig, um sie unter dem Mikroskop vollständig zu erfassen. Heute lesen Forschende häufig ihre DNA, um zu erkennen, welche Mikroben vorhanden sind und welche Funktionen sie haben könnten. Mehr DNA‑Daten bedeuten jedoch höhere Kosten. Diese Studie stellt eine einfache, aber wichtige Frage: Wie viel DNA‑Sequenzierung ist tatsächlich nötig, um nützliche Antworten über eine mikrobielle Gemeinschaft zu erhalten, und ab wann führt Sparen in die Irre?
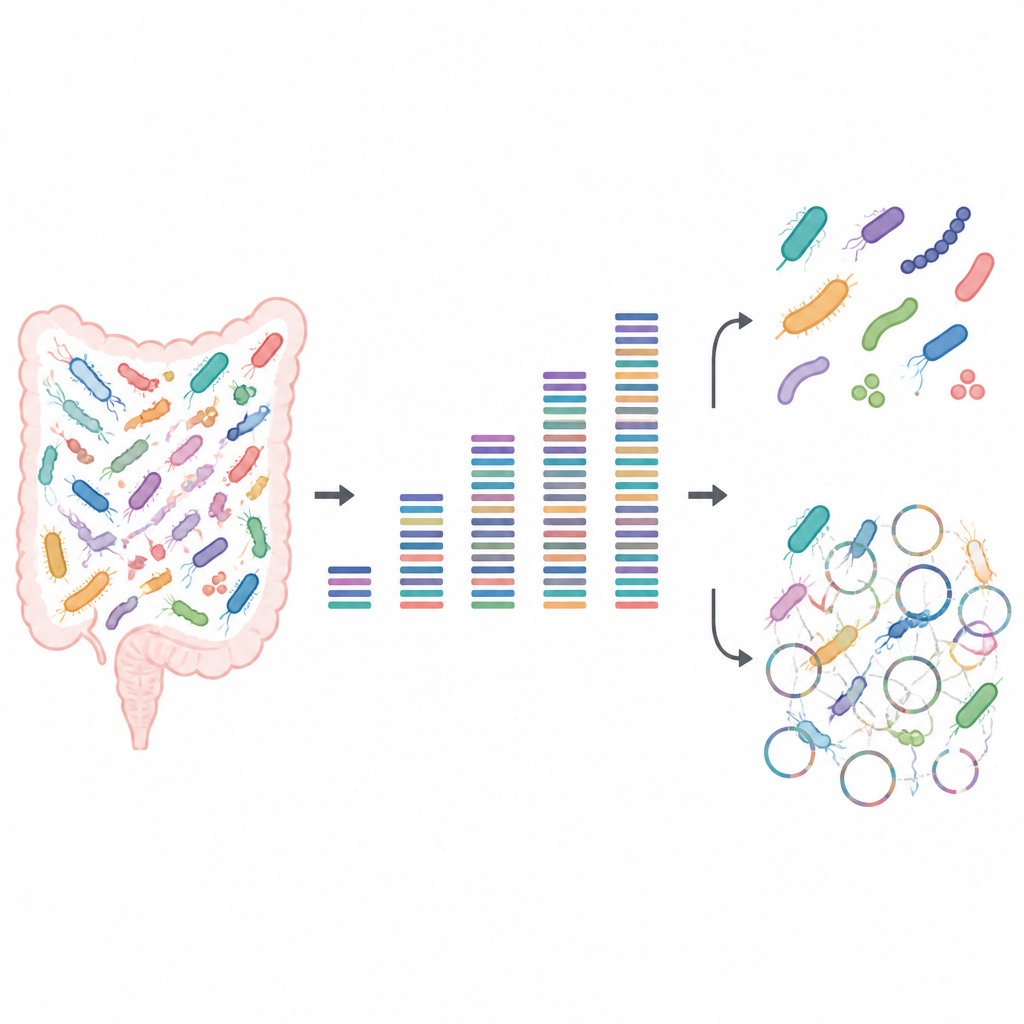
Die Leistungsfähigkeit von kleinen und großen Datensätzen testen
Die Forschenden bauten künstliche mikrobielle Gemeinschaften aus bekannten Darmbakterien, die im Labor kultiviert wurden. Da sie genau wussten, welche Stämme in welchen Mengen vorhanden waren, dienten diese „Mock“-Proben wie ein Testbild eines Fernsehers und zeigten, wo das Bild der DNA‑Sequenzierung scharf oder verschwommen war. Sie sequenzierten jede Gemeinschaft in Tiefen von sehr wenig bis sehr viel Daten und analysierten dann, welche Bakterien nachgewiesen werden konnten, ob nah verwandte Stämme voneinander zu unterscheiden waren und wie viel vom proteinbildenden Potenzial jedes Stamms wiederhergestellt werden konnte.
Was mit wenig Sequenzierung gut funktioniert
Für grundlegende Fragen wie „Welche Arten sind hier?“ und „Wie häufig ist jede einzelne?“ fanden die Autorinnen und Autoren, dass überraschend wenig Daten nötig sind, sofern gute Referenzgenome verfügbar sind. Selbst bei geringer Sequenzierung hinterließ jeder Stamm eine nachweisbare Spur, und die Muster der relativen Häufigkeit blieben stabil, wenn mehr Daten hinzukamen. Etwa eine halbe Gigabase DNA pro Probe reichte aus, um die Zusammensetzung der Gemeinschaft zuverlässig zu erfassen. Das macht flache Sequenzierung attraktiv für große Studien, die Gesamtmuster des Mikrobioms über viele Personen oder Bedingungen hinweg vergleichen wollen, ohne ein Vermögen auszugeben.
Worin flache Ansätze versagen
Probleme traten auf, sobald der Fokus von Arten auf einzelne Stämme und deren detaillierte Funktionen verschoben wurde. Das Rekonstruieren ganzer Genome de novo, ein Prozess, der als Metagenom-Assembly bezeichnet wird, benötigte deutlich tiefere Sequenzierung und scheiterte dennoch oft. Software gruppierte DNA‑Fragmente zu Entwürfen von Genomen, die nach Standardprüfungen hochwertig aussahen, doch viele dieser Entwürfe waren tatsächlich Flickwerke aus mehreren verschiedenen Stämmen. Selbst bei sehr hoher Sequenzierungstiefe war ein nennenswerter Anteil der assemblierten Genome chimärisch, und einige echte Stämme wurden ganz übersehen. Flache Sequenzierung hatte außerdem Schwierigkeiten, das vollständige Set an Proteinen zu erfassen: Während wenige Gigabases ausreichten, um grobe Stoffwechselwege zu skizzieren, war deutlich tiefere Sequenzierung erforderlich, um die meisten einzelnen Proteine abzudecken, insbesondere in komplexeren Gemeinschaften.
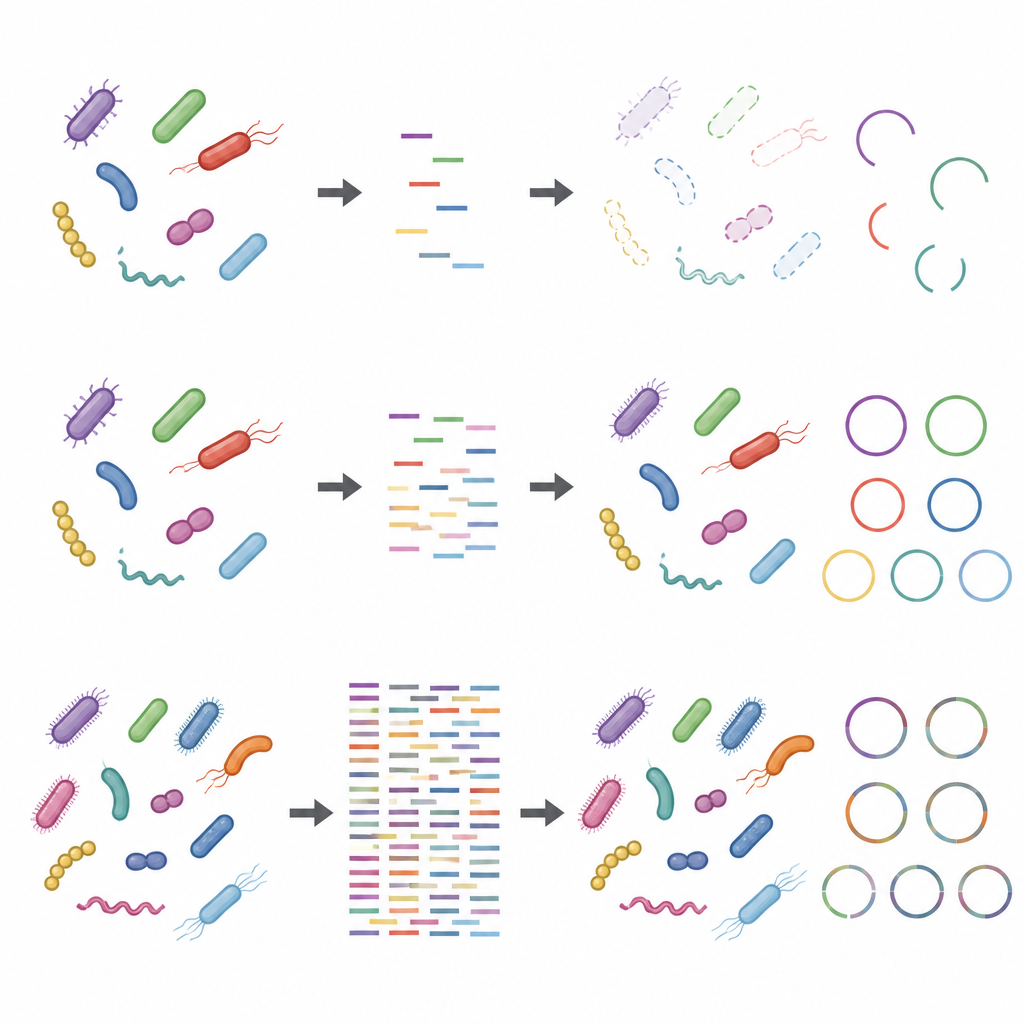
Einfluss von Laborentscheidungen und Fremd‑DNA
Die Studie zeigte außerdem, dass Schritte vor dem Einbringen der DNA in den Sequencer die Ergebnisse verzerren können, besonders bei geringer Sequenzierungstiefe. Mehr Ausgangs‑DNA und weniger Amplifikationszyklen machten taxonomische und funktionelle Profile robuster. Dagegen verzerrten Protokolle mit wenig Input‑DNA und vielen Amplifikationszyklen die relativen Häufigkeiten einiger Stämme. Das Hinzufügen von Wirts‑DNA, das Proben nachahmt, die reich an humanem oder tierischem Material sind, verringerte zusätzlich die scheinbare Abdeckung mikrobieller Genome und ihrer Proteine. Diese Effekte wurden bei höherer Sequenzierungstiefe weniger gravierend, verschwanden aber nicht vollständig.
Praktische Empfehlungen für künftige Mikrobiom‑Studien
Insgesamt liefert die Arbeit einen Realitätscheck darüber, was flache DNA‑Sequenzierung leisten kann und was nicht. Für Studien, die hauptsächlich eine grobe Bestandsaufnahme darüber benötigen, welche Mikroben in gut untersuchten Umgebungen wie dem menschlichen Darm vorkommen, können moderate Sequenzierungstiefen gut funktionieren, sofern gute Referenzgenome vorhanden sind und die Laborprotokolle sorgfältig gewählt werden. Für detaillierte Fragen zu Funktionen einer Gemeinschaft oder feinen Unterschieden zwischen Stämmen reicht flache Sequenzierung jedoch nicht aus. Selbst sehr tiefe Sequenzierung kann das Problem der vermischten assemblierten Genome nicht vollständig beheben; Ergebnisse, die auf solchen Entwürfen beruhen, sollten daher mit Vorsicht interpretiert werden. Kurz gesagt: Menge der DNA‑Daten und Analysemethoden sollten an die wissenschaftliche Fragestellung angepasst werden, mit klarem Bewusstsein dafür, wo das Bild unscharf bleibt.
Zitation: Treichel, N.S., Pauvert, C., Séneca, J. et al. Benchmarking of shotgun sequencing depth reveals the potential and limitations of shallow metagenomics and strain-level analysis. Nat Microbiol 11, 1233–1244 (2026). https://doi.org/10.1038/s41564-026-02334-2
Schlüsselwörter: Microbiom-Sequenzierung, flache Metagenomik, Sequenzierungstiefe, metagenomisch assemblierte Genome, Analyse auf Stammebene