Clear Sky Science · fr
Évaluation de la profondeur de séquençage shotgun révèle le potentiel et les limites de la métagénomique peu profonde et de l’analyse au niveau des souches
Pourquoi observer le monde microscopique nécessite la bonne quantité de données
Les microbes qui vivent en nous et sur nous influencent notre santé, mais ils sont trop petits et divers pour être comptés au microscope. Aujourd’hui, les chercheurs lisent souvent leur ADN pour savoir quelles espèces sont présentes et quelles fonctions elles peuvent assurer. Mais davantage de données d’ADN signifie des coûts plus élevés. Cette étude pose une question simple mais cruciale : quelle quantité de séquençage est réellement nécessaire pour obtenir des réponses utiles sur une communauté microbienne, et à quel point réduire les coûts commence-t-il à induire en erreur ?
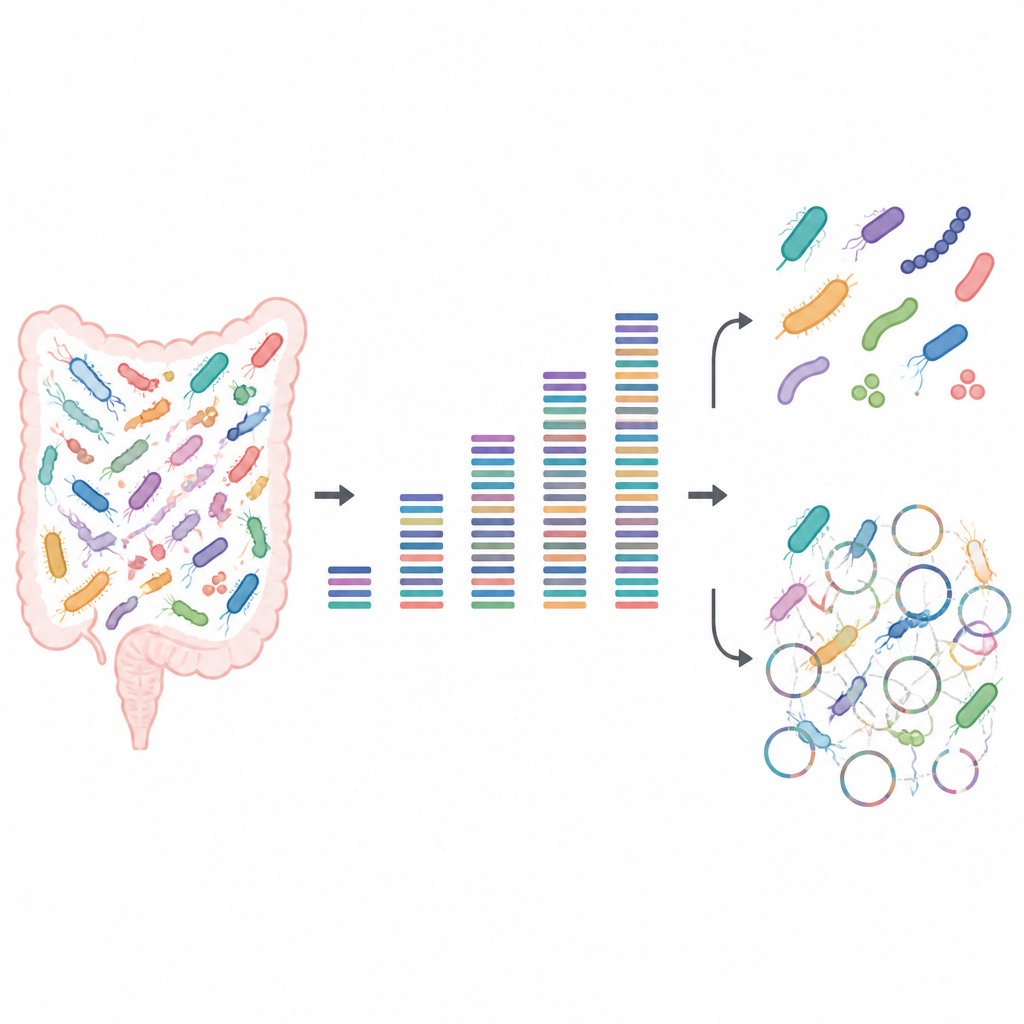
Tester la puissance des petites et grandes quantités de données
Les chercheurs ont construit des communautés microbiennes artificielles à partir de bactéries intestinales connues cultivées en laboratoire. Comme ils savaient exactement quelles souches étaient présentes et en quelles proportions, ces échantillons « factices » ont servi de motif test, révélant où l’image produite par le séquençage ADN était nette ou floue. Ils ont séquencé chaque communauté à des profondeurs allant de très faibles à très élevées, puis ont analysé quelles bactéries étaient détectables, si des souches proches pouvaient être distinguées, et quelle part du potentiel de production protéique de chaque souche pouvait être récupérée.
Ce qui fonctionne bien avec peu de séquençage
Pour des questions basiques comme « quelles espèces sont présentes » et « quelle est la fréquence relative de chacune », l’équipe a constaté qu’assez peu de données suffisaient lorsque de bons génomes de référence étaient disponibles. Même à faible profondeur de séquençage, chaque souche laissait une trace détectable, et les profils d’abondance relative restaient stables à mesure que plus de données étaient ajoutées. Environ une demi-gigabase d’ADN par échantillon a été suffisante pour établir de manière fiable la composition de la communauté. Cela rend le séquençage peu profond intéressant pour les grandes études qui veulent comparer des tendances globales du microbiome entre de nombreuses personnes ou conditions sans dépenser une fortune.
Où les approches peu profondes montrent leurs limites
Des problèmes sont apparus lorsque l’attention s’est déplacée des espèces vers les souches individuelles et leurs fonctions détaillées. Reconstruire des génomes complets à partir de zéro, un processus appelé assemblage métagénomique, nécessitait un séquençage beaucoup plus profond et échouait encore souvent. Les programmes informatiques regroupaient des fragments d’ADN en génomes provisoires qui semblaient de haute qualité selon les listes de contrôle habituelles, mais dont beaucoup étaient en réalité des patchworks composés de plusieurs souches différentes. Même à des profondeurs de séquençage très élevées, une part notable de ces génomes assemblés était chimérique, et certaines souches véritables étaient complètement manquées. Le séquençage peu profond avait aussi du mal à capturer l’ensemble des protéines présentes : quelques gigabases suffisaient pour tracer les grandes voies métaboliques, mais un séquençage beaucoup plus profond était nécessaire pour couvrir la plupart des protéines individuelles, en particulier dans les communautés plus complexes.
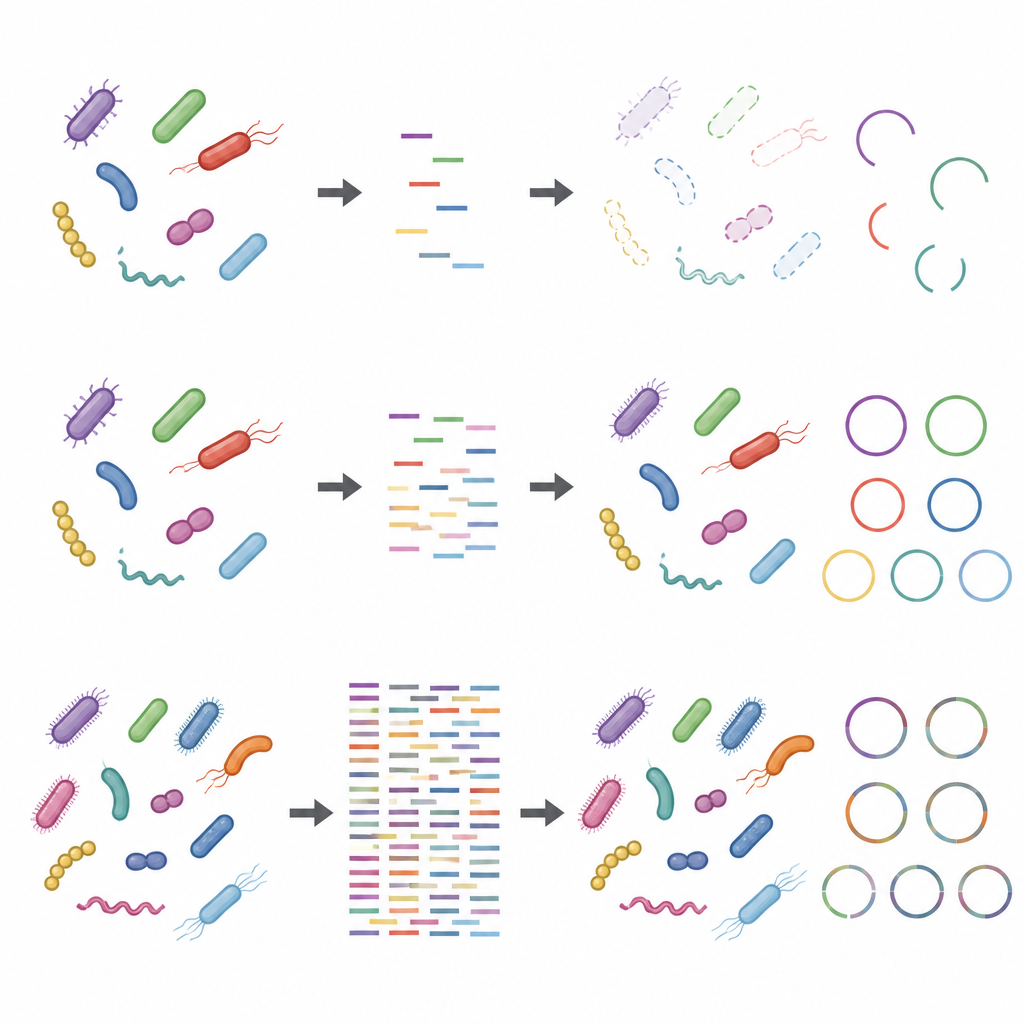
Effets des choix en laboratoire et de l’ADN parasite
L’étude a également montré que les étapes réalisées avant que l’ADN n’atteigne le séquenceur peuvent biaiser les résultats, en particulier lorsque la profondeur de séquençage est faible. Utiliser davantage d’ADN de départ et réduire le nombre de cycles d’amplification rendait les profils taxonomiques et fonctionnels plus robustes. En revanche, des protocoles avec peu d’ADN initial et de nombreux cycles d’amplification déformaient l’abondance relative de certaines souches. L’ajout d’ADN hôte, simulant ce qui se produit avec des échantillons riches en matériel humain ou animal, réduisait encore la couverture apparente des génomes microbiennes et de leurs protéines. Ces problèmes s’atténuaient à des profondeurs de séquençage plus élevées, mais ne disparaissaient pas complètement.
Conseils pratiques pour les futures études du microbiome
Globalement, ce travail fournit un examen réaliste de ce que le séquençage ADN peu profond peut et ne peut pas fournir. Pour les études qui ont principalement besoin d’un recensement large des microbes présents dans un environnement bien étudié comme l’intestin humain, des profondeurs de séquençage modestes peuvent suffire, à condition que de bons génomes de référence existent et que les protocoles de laboratoire soient soigneusement choisis. En revanche, pour des questions détaillées sur les fonctions d’une communauté ou les différences fines entre souches, le séquençage peu profond n’est pas adéquat. Même un séquençage très profond ne résout pas complètement le problème des génomes assemblés mélangés, aussi les résultats basés sur de tels brouillons doivent être interprétés avec prudence. En bref, la quantité de données d’ADN et les méthodes d’analyse doivent être adaptées à la question scientifique, en ayant une compréhension claire des zones où l’image reste floue.
Citation: Treichel, N.S., Pauvert, C., Séneca, J. et al. Benchmarking of shotgun sequencing depth reveals the potential and limitations of shallow metagenomics and strain-level analysis. Nat Microbiol 11, 1233–1244 (2026). https://doi.org/10.1038/s41564-026-02334-2
Mots-clés: séquencage du microbiome, métagénomique peu profonde, profondeur de séquençage, génomes assemblés à partir du métagénome, analyse au niveau des souches