Clear Sky Science · sv
Inkorporera långväga interaktioner via multipolutveckling i mark- och exciterade tillstånds molekylsimuleringar
Varför avlägsna krafter spelar roll i kemi
Många av de viktigaste processerna inom kemi och biologi – från hur läkemedel binder till proteiner till hur solceller fångar ljus – beror på subtila elektriska krafter som verkar över förvånansvärt långa avstånd. Att simulera dessa effekter korrekt har traditionellt krävt mycket kostsamma kvantmekaniska beräkningar, vilket begränsar storleken på system och tidsskalor som forskare kan studera. Den här artikeln introducerar ett nytt maskininlärningssätt, kallat Field-MACE, som gör det mycket effektivare att inkludera dessa långväga krafter vid simulering av molekyler i komplexa miljöer som vätskor och biologiskt material.
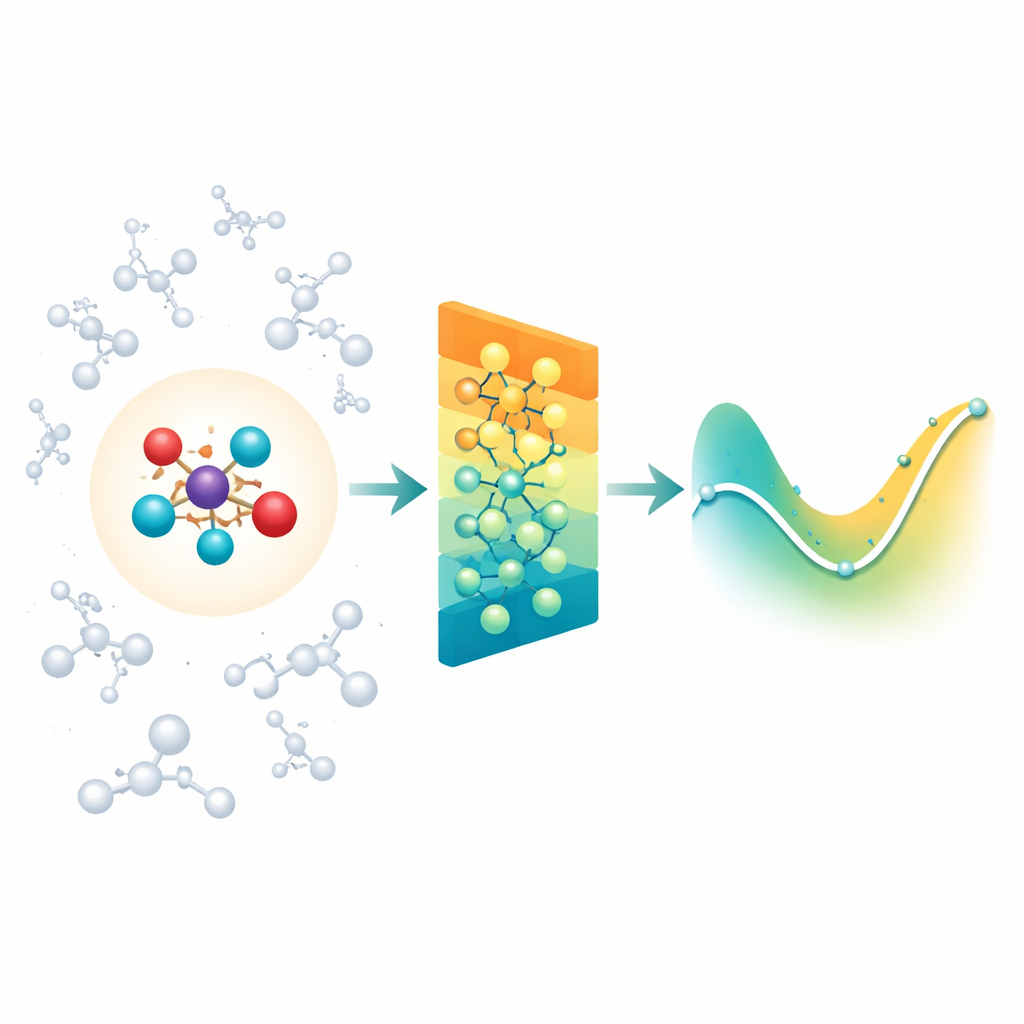
Blanda detaljerad kemi med dess omgivning
När kemister simulerar reaktioner i lösning eller inne i proteiner använder de ofta ett "inzoomat" kvantområde där bindningar bryts och bildas, omgivet av ett större "klassiskt" område som utgör miljön. Svårigheten är att miljön fortfarande utövar långväga elektriska krafter på den reaktiva kärnan, och de flesta moderna neurala nätverksmodeller för molekyler fokuserar främst på interaktioner mellan närliggande grannar. Som ett resultat kan de missa viktiga lösningsmedels- eller proteineffekter, eller bli opraktiskt långsamma om de försöker räkna varje avlägsen interaktion en och en. Författarna tar itu med detta genom att bygga vidare på en befintlig klass av rotationsmedvetna neurala nätverk (MACE) och utvidga dem så att kvantregionen effektivt kan känna påverkan från ett stort moln av klassiska laddningar runt den.
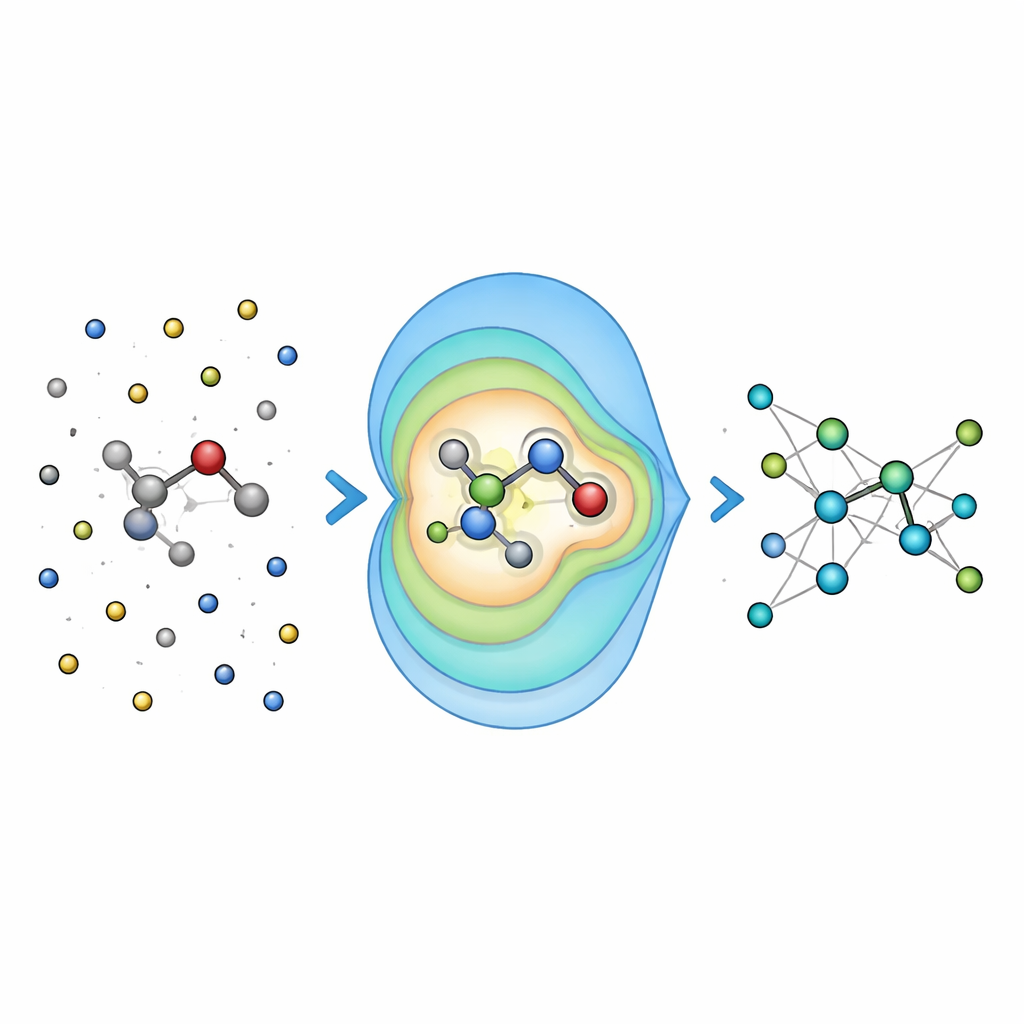
Sammanfatta avlägsna laddningar till enkla mönster
I stället för att mata nätverket med varje enskild interaktion mellan omgivningen och varje atom använder Field-MACE ett klassiskt fysiktrick känt som multipolutveckling. Enkelt uttryckt komprimerar detta påverkan från många laddningar till ett litet antal riktade mönster: total laddning, genomsnittlig orientering av det elektriska fältet och mer förfinade former som beskriver hur fältet böjer sig i rummet. Dessa mönster uttrycks naturligt som vinkelformer runt molekylen och harmoniserar med hur det underliggande neurala nätverket representerar geometri. Modellen kombinerar två informationsströmmar under inlärning: lokala, kortdistansmeddelanden som beskriver bindningar och närliggande grannar, och långdistansmeddelanden byggda från dessa multipolmönster som sammanfattar hur omgivande lösningsmedel eller material drar i kvantregionen.
Testa noggrannheten på verkliga kemiska problem
För att se om idén betalade sig utvärderade forskarna först Field-MACE på molekyler upplösta i vätskor och jämförde det med mer traditionella sätt att hantera långväga elektrostatik, såsom Ewald-summation och enkla Coulombpotentialer. De fann att inkludering av ett måttligt antal multipoltermer dramatiskt förbättrade noggrannheten i predicerade energier och krafter, särskilt för större och mer flexibla molekyler vars former starkt påverkas av deras lösningsmedel. Avgörande var att denna noggrannhetsökning gavs med endast en liten ökning i beräkningskostnad, eftersom den dyra delen av beräkningen fortfarande i huvudsak skalar med storleken på kvantkärnan, inte med havet av omkringliggande atomer.
Från metalkatalysatorer till ljusstyrda reaktioner
Teamet applicerade sedan Field-MACE på två krävande fallstudier. I det första modellerade de nickelbaserade katalysatorer i flytande bensen, en klass av metallkomplex viktiga i många industriella reaktioner. Den nya modellen gav stabil molekylär dynamik över många pikosekunder och reproducerade viktiga strukturella egenskaper, såsom bindningslängder och vinklar, även för komplex som inte uttryckligen ingick i träningen. I det andra fallet studerade de en fotokemisk "ringöppnings"-reaktion av den lilla molekylen furan i vatten, en process som involverar exciterade elektroniska tillstånd och snabba hopp däremellan. Trots komplexiteten i dessa ljusstyrda dynamiker matchade Field-MACE i hög grad högklassiga kvantsimuleringar vad gäller hur populationen av olika elektroniska tillstånd utvecklades över tid.
Lära snabbare genom att återanvända kemisk kunskap
En stor praktisk utmaning i sådana simuleringar är knappheten och kostnaden för referensdata som redan inkluderar miljöeffekter. Författarna visade att de dramatiskt kan minska denna kostnad genom att börja från en förtränad grundmodell som ursprungligen tränats endast på isolerade molekyler utan lösningsmedel. Genom att finjustera denna befintliga "kortdistans"-kunskap och lägga till det multipolbaserade långdistanslagret nådde Field-MACE hög noggrannhet med betydligt färre datapunkter. Detta var särskilt påtagligt för exciterad-tillstånds dynamik hos furan: när endast några dussin dyra referensberäkningar fanns tillgängliga misslyckades modeller tränade från början, medan de som initierats från grundmodellen fortfarande fångade den väsentliga reaktionsvägen.
Vad detta innebär för framtida simuleringar
I vardagliga termer ger Field-MACE kemister och materialforskare ett sätt att behålla den detaljerade kvantsynen där den är viktig samtidigt som man känner dragningen från en stor, komplex omgivning – allt detta utan att betala det vanliga beräkningspriset. Genom att komprimera avlägsna elektriska effekter till en kompakt uppsättning mönster och kombinera dem med kraftfulla neurala nätverk möjliggör metoden noggranna, skalbara simuleringar för både grundtillstånd och exciterade tillstånd. Detta öppnar dörren för att studera mer realistiska system – från metalkatalysatorer i lösning till fotoaktiva molekyler i biologiska miljöer – och att göra det med avsevärt mindre träningsdata än tidigare.
Citering: Barrett, R., Dietschreit, J.C.B. & Westermayr, J. Incorporating long-range interactions via the multipole expansion into ground and excited-state molecular simulations. npj Comput Mater 12, 135 (2026). https://doi.org/10.1038/s41524-026-02048-3
Nyckelord: maskininlärningspotentialer, kvantmekanik/molekylmekanik, långväga elektrostatik, molekylsimulationer, exciterade-tillstånds dynamik