Clear Sky Science · it
Incorporare interazioni a lungo raggio tramite l’espansione multipolare nelle simulazioni molecolari di stati fondamentali ed eccitati
Perché le forze distanti contano in chimica
Molti dei processi più importanti in chimica e biologia – da come i farmaci si legano alle proteine a come le celle solari raccolgono la luce – dipendono da sottili forze elettriche che agiscono su distanze sorprendentemente grandi. Simulare accuratamente questi effetti ha tradizionalmente richiesto calcoli quantistici molto costosi, il che limita le dimensioni dei sistemi e le scale temporali che gli scienziati possono studiare. Questo articolo introduce un nuovo approccio di apprendimento automatico, chiamato Field-MACE, che rende molto più efficiente l’inclusione di tali forze a lungo raggio nella simulazione di molecole in ambienti complessi come liquidi e materia biologica.
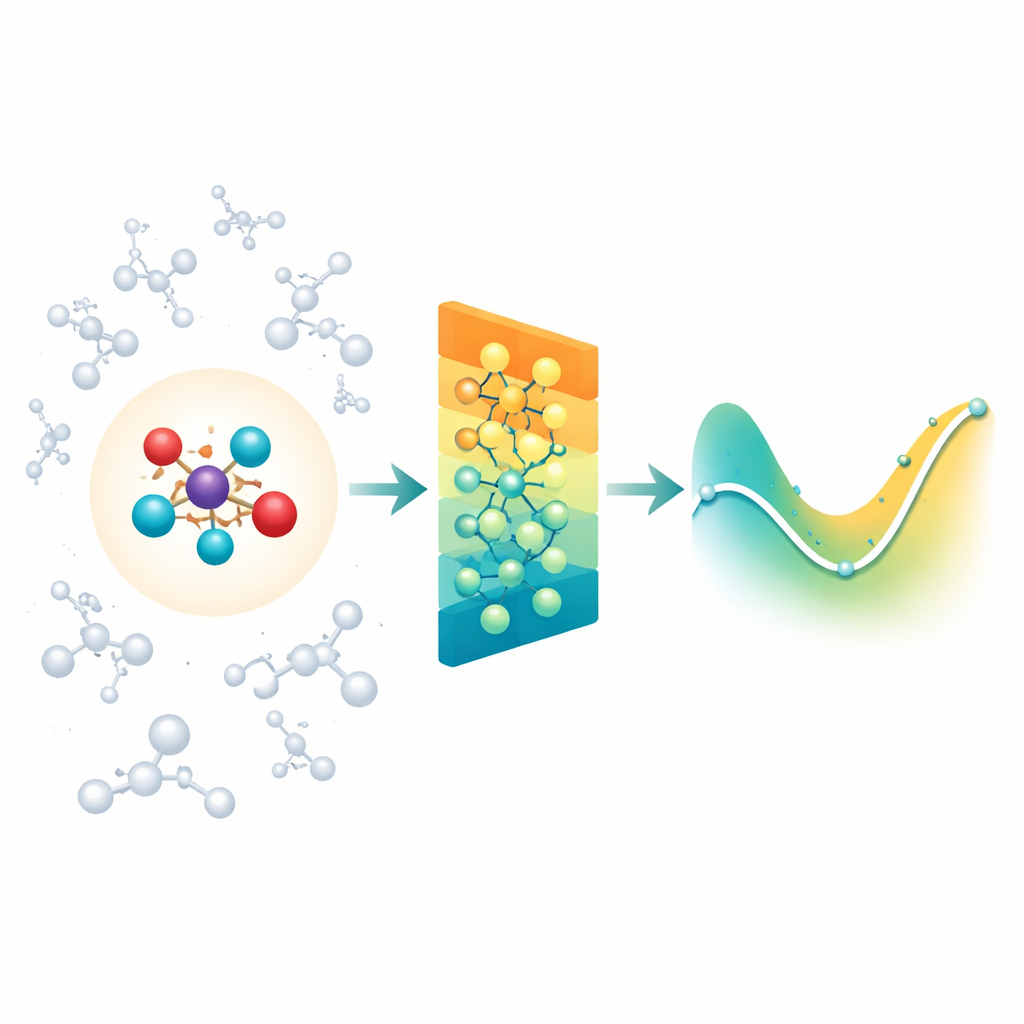
Fondere la chimica dettagliata con l’ambiente circostante
Quando i chimici simulano reazioni in soluzione o all’interno di proteine, spesso usano una regione quantistica “ingrandita” dove legami si spezzano e si formano, circondata da una più ampia regione “classica” che fornisce l’ambiente. La difficoltà è che l’ambiente esercita comunque forze elettriche a lungo raggio sul nucleo reattivo, e la maggior parte dei modelli basati su reti neurali per molecole si concentra principalmente sulle interazioni tra vicini prossimi. Di conseguenza, possono perdere effetti importanti del solvente o della proteina, o diventare proibitivamente lenti se cercano di contabilizzare ogni interazione distante una per una. Gli autori affrontano questo problema sviluppando una classe esistente di reti neurali consapevoli della rotazione (MACE) e estendendola in modo che la regione quantistica possa percepire in modo efficiente l’influenza di una vasta nube di cariche classiche attorno a essa.
Riassumere cariche lontane in schemi semplici
Invece di fornire alla rete ogni singola interazione tra l’ambiente e ciascun atomo, Field-MACE utilizza un trucco classico della fisica noto come espansione multipolare. In termini semplici, questo comprime l’influenza di molte cariche in un piccolo insieme di schemi direzionali: carica complessiva, orientamento medio del campo elettrico e forme più raffinate che descrivono come il campo curva nello spazio. Questi schemi si esprimono naturalmente come forme angolari attorno alla molecola e si integrano con il modo in cui la rete neurale sottostante rappresenta la geometria. Il modello combina due flussi di informazione durante l’apprendimento: messaggi locali e a breve raggio che descrivono legami e vicini prossimi, e messaggi a lungo raggio costruiti a partire da questi pattern multipolari che riassumono come il solvente o il materiale circostante tira sul nucleo quantistico.
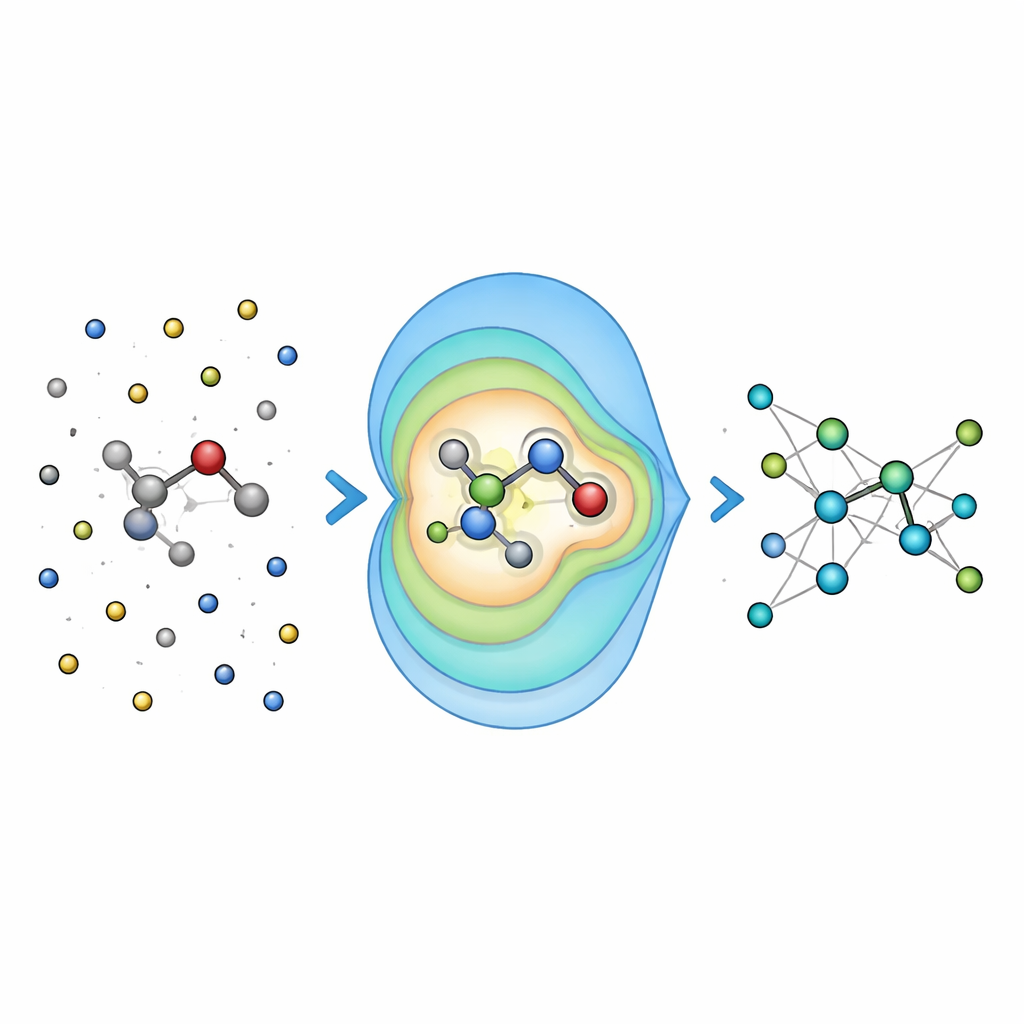
Verificare l’accuratezza su problemi chimici reali
Per capire se l’idea funziona, i ricercatori hanno prima valutato Field-MACE su molecole disciolte in liquidi, confrontandolo con modalità più tradizionali di trattare l’elettrostatica a lungo raggio, come la sommatoria di Ewald e i semplici potenziali coulombiani. Hanno trovato che includere un numero modesto di termini multipolari migliorava drasticamente l’accuratezza delle energie e delle forze previste, in particolare per molecole più grandi e flessibili le cui forme sono fortemente influenzate dal solvente. Crucialmente, questo aumento di accuratezza è avvenuto con solo un piccolo incremento del costo computazionale, perché la parte costosa del calcolo scala ancora principalmente con la dimensione del nucleo quantistico, non con il mare di atomi circostanti.
Da catalizzatori metallici a reazioni indotte dalla luce
Il team ha poi applicato Field-MACE a due studi di caso esigenti. Nel primo, hanno modellato catalizzatori a base di nichel in benzene liquido, una classe di complessi metallici importante in molte reazioni industriali. Il nuovo modello ha prodotto dinamiche molecolari stabili per molti picosecondi e ha riprodotto caratteristiche strutturali chiave, come lunghezze di legame e angoli, anche per complessi non inclusi esplicitamente nel training. Nel secondo caso, hanno esaminato una reazione fotochimica di “apertura dell’anello” della piccola molecola furano in acqua, un processo che coinvolge stati elettronici eccitati e rapidi salti tra di essi. Nonostante la complessità di queste dinamiche indotte dalla luce, Field-MACE ha riprodotto da vicino le simulazioni quantistiche di alto livello nell’evoluzione temporale della popolazione dei diversi stati elettronici.
Imparare più in fretta riutilizzando conoscenza chimica
Un ostacolo pratico importante in tali simulazioni è la scarsità e il costo dei dati di riferimento che includono già gli effetti ambientali. Gli autori hanno dimostrato di poter ridurre drasticamente questo costo partendo da un modello di base pre-addestrato che era stato originariamente allenato solo su molecole isolate senza solvente. Affinando questa conoscenza “a corto raggio” esistente e aggiungendo lo strato a lungo raggio basato sui multipoli, Field-MACE ha raggiunto alta accuratezza con molti meno punti dati. Questo è stato particolarmente evidente per le dinamiche di stati eccitati del furano: quando erano disponibili solo poche dozzine di costose valutazioni di riferimento, i modelli addestrati da zero fallivano, mentre quelli inizializzati dal modello di base catturavano comunque il percorso reattivo essenziale.
Cosa significa per le simulazioni future
In termini pratici, Field-MACE offre a chimici e scienziati dei materiali un modo per mantenere la vista quantistica dettagliata dove è importante, pur percependo la spinta di un ambiente ampio e complesso – il tutto senza pagare il solito prezzo computazionale. Comprendo gli effetti elettrici lontani in un compatto insieme di schemi e combinandoli con potenti reti neurali, il metodo consente simulazioni accurate e scalabili sia per stati fondamentali che eccitati. Questo apre la porta allo studio di sistemi più realistici – dai catalizzatori metallici in soluzione alle molecole fotoattive in contesti biologici – e a farlo con molti meno dati di addestramento rispetto al passato.
Citazione: Barrett, R., Dietschreit, J.C.B. & Westermayr, J. Incorporating long-range interactions via the multipole expansion into ground and excited-state molecular simulations. npj Comput Mater 12, 135 (2026). https://doi.org/10.1038/s41524-026-02048-3
Parole chiave: potenziali di apprendimento automatico, meccanica quantistica/meccanica molecolare, elettrostatica a lungo raggio, simulazioni molecolari, dynamics di stati eccitati