Clear Sky Science · de
Einbeziehung von Fernwechselwirkungen durch Multipolentwicklung in Grundzustands- und angeregten molekularen Simulationen
Warum entfernte Kräfte in der Chemie wichtig sind
Viele der wichtigsten Prozesse in Chemie und Biologie – von der Bindung von Wirkstoffen an Proteine bis zur Lichtnutzung in Solarzellen – hängen von feinen elektrischen Kräften ab, die über überraschend große Entfernungen wirken. Diese Effekte genau zu simulieren erforderte bislang sehr aufwändige quantenmechanische Rechnungen, was die Größe der untersuchbaren Systeme und die möglichen Zeitskalen begrenzt. Diese Arbeit stellt einen neuen maschinellen Lernansatz vor, Field-MACE genannt, der es deutlich effizienter macht, diese langreichweitigen Kräfte bei Simulationen von Molekülen in komplexen Umgebungen wie Flüssigkeiten oder biologischer Materie zu berücksichtigen.
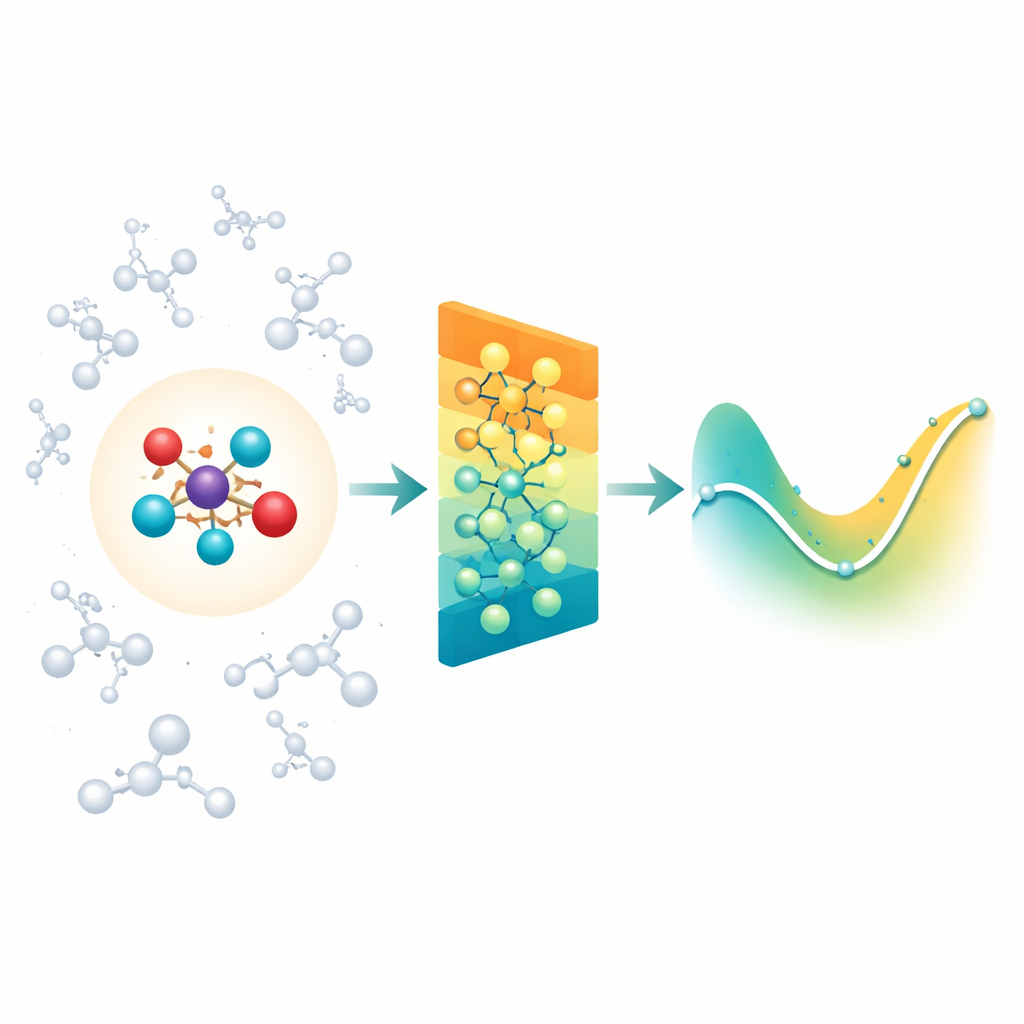
Die detaillierte Chemie mit ihrer Umgebung verbinden
Wenn Chemiker Reaktionen in Lösung oder innerhalb von Proteinen simulieren, verwenden sie oft eine „hineingezoomte“ Quantenregion, in der Bindungen brechen und entstehen, umgeben von einer größeren „klassischen“ Region, die die Umgebung liefert. Das Problem ist, dass die Umgebung weiterhin langreichweitige elektrische Kräfte auf den reaktiven Kern ausübt, und die meisten modernen neuronalen Netze für Moleküle konzentrieren sich hauptsächlich auf Wechselwirkungen zwischen nahen Nachbarn. Dadurch können wichtige Lösungsmittel- oder Proteineffekte übersehen werden oder die Modelle werden unpraktikabel langsam, wenn sie versuchen, jede entfernte Wechselwirkung einzeln zu berücksichtigen. Die Autoren gehen dieses Problem an, indem sie auf einer bestehenden Klasse rotationssensitiver neuronaler Netze (MACE) aufbauen und diese so erweitern, dass die Quantenregion effizient den Einfluss einer großen Wolke klassischer Ladungen um sie herum spüren kann.
Fernen Ladungen in einfache Muster zusammenfassen
Anstatt dem Netzwerk jede einzelne Wechselwirkung zwischen Umgebung und jedem Atom zuzuführen, nutzt Field-MACE einen klassischen physikalischen Trick, die Multipolentwicklung. Vereinfacht gesagt komprimiert diese Methode den Einfluss vieler Ladungen in eine kleine Menge richtungsabhängiger Muster: die Gesamtladung, die mittlere Orientierung des elektrischen Feldes und feinere Formen, die beschreiben, wie sich das Feld im Raum krümmt. Diese Muster lassen sich natürlich als Winkelmuster um das Molekül ausdrücken und fügen sich gut in die geometrische Repräsentation des zugrundeliegenden neuronalen Netzes ein. Das Modell kombiniert während des Lernens zwei Informationsströme: lokale, kurzreichweitige Botschaften, die Bindungen und nahe Nachbarn beschreiben, und langreichweitige Botschaften, die aus diesen Multipolmuster aufgebaut sind und zusammenfassen, wie das umgebende Lösungsmittel oder Material an der Quantenregion zieht.
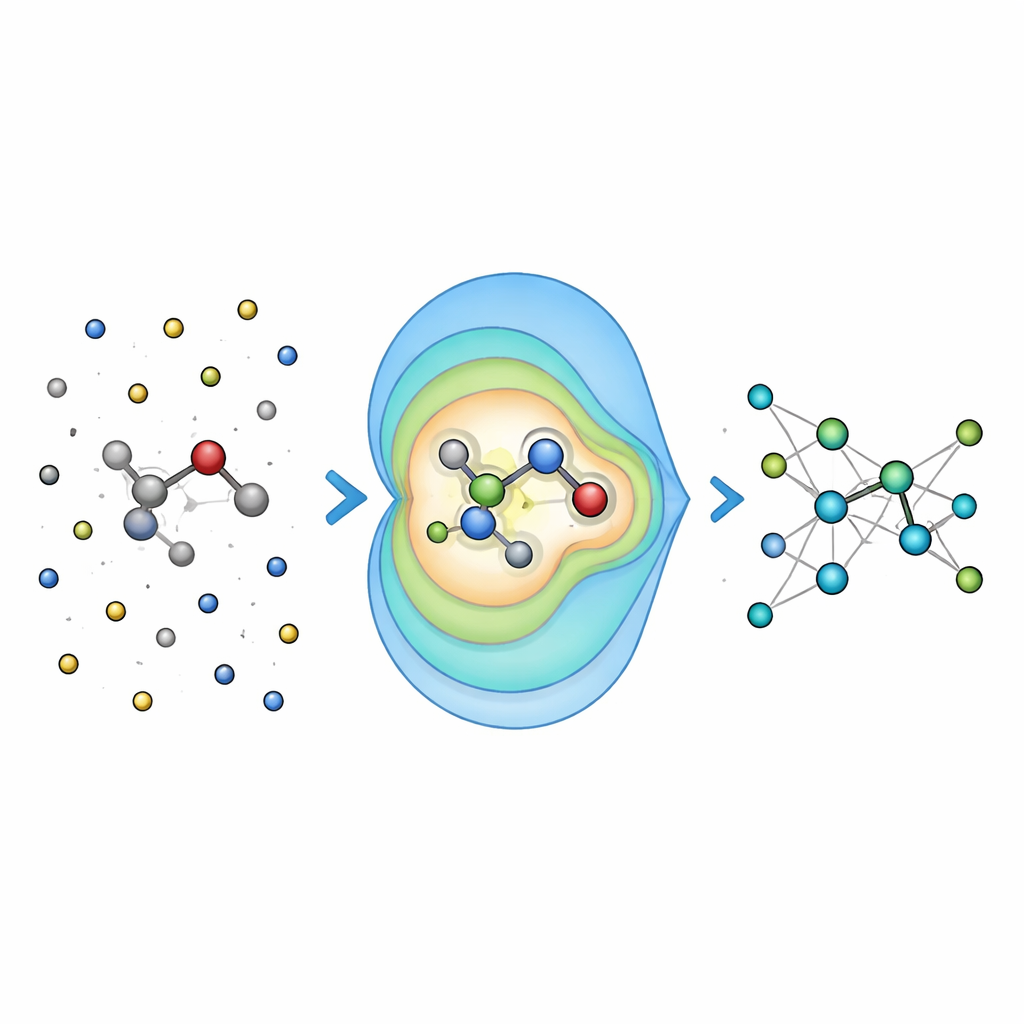
Genauigkeit an realen chemischen Problemen testen
Um zu prüfen, ob diese Idee sich auszahlt, testeten die Forscher Field-MACE zunächst an in Flüssigkeiten gelösten Molekülen und verglichen es mit traditionelleren Methoden zur Behandlung langreichweitiger Elektrostatiek, wie der Ewald-Summe und einfachen Coulomb-Potentialen. Sie fanden heraus, dass die Aufnahme einer moderaten Anzahl von Multipoltermen die Vorhersagegenauigkeit für Energien und Kräfte drastisch verbesserte, insbesondere für größere und flexiblere Moleküle, deren Form stark durch das Lösungsmittel beeinflusst wird. Entscheidend war, dass dieser Genauigkeitsgewinn nur mit einem geringen Mehraufwand an Rechenzeit einherging, weil der teure Teil der Rechnung weiterhin hauptsächlich mit der Größe des Quantenkerns skaliert und nicht mit dem Meer der umgebenden Atome.
Von Metallkatalysatoren bis zu lichtgetriebenen Reaktionen
Das Team wandte Field-MACE anschließend auf zwei anspruchsvolle Fallstudien an. Zunächst modellierten sie nickelbasierte Katalysatoren in flüssigem Benzol, eine Klasse von Metallkomplexen, die in vielen industriellen Reaktionen wichtig sind. Das neue Modell erzeugte stabile Molekulardynamiken über viele Pikosekunden und reproduzierte wichtige Strukturmerkmale wie Bindungslängen und Winkel, selbst für Komplexe, die nicht explizit im Training enthalten waren. Im zweiten Fall betrachteten sie eine photochemische Ringöffnungsreaktion des kleinen Moleküls Furans in Wasser, einen Prozess, der angeregte elektronische Zustände und schnelle Übergänge zwischen ihnen umfasst. Trotz der Komplexität dieser lichtgetriebenen Dynamik stimmte Field-MACE eng mit hochrangigen Quantenberechnungen überein, was die zeitliche Entwicklung der Populationen verschiedener elektronischer Zustände betrifft.
Schneller lernen durch Wiederverwendung chemischen Wissens
Ein praktisches Hindernis bei solchen Simulationen ist die Knappheit und der hohe Preis von Referenzdaten, die Umwelteffekte bereits enthalten. Die Autoren zeigten, dass sich diese Kosten drastisch senken lassen, wenn man von einem vortrainierten Basismodell ausgeht, das ursprünglich nur an isolierten Molekülen ohne Lösungsmittel trainiert wurde. Durch Feinabstimmung dieses vorhandenen „kurzreichweitigen“ Wissens und das Hinzufügen der multipolbasierten langreichweitigen Schicht erreichte Field-MACE hohe Genauigkeit mit deutlich weniger Datenpunkten. Besonders eindrucksvoll war dies bei der angeregten Zustandsdynamik von Furan: Als nur einige Dutzend teurer Referenzrechnungen verfügbar waren, scheiterten Modelle, die von Grund auf neu trainiert wurden, während solche, die vom Basismodell aus gestartet waren, den wesentlichen Reaktionsweg erfassten.
Was das für zukünftige Simulationen bedeutet
Alltagsmäßig gesagt bietet Field-MACE Chemikern und Materialwissenschaftlern eine Möglichkeit, die detaillierte Quantenperspektive dort beizubehalten, wo sie gebraucht wird, und gleichzeitig die Einflüsse einer großen, komplexen Umgebung zu spüren – und das ohne den üblichen Rechenaufwand. Indem ferne elektrische Effekte in ein kompaktes Set von Mustern komprimiert und mit leistungsfähigen neuronalen Netzen kombiniert werden, ermöglicht die Methode genaue, skalierbare Simulationen sowohl für Grund- als auch für angeregte Zustände. Das eröffnet die Möglichkeit, realistischere Systeme zu untersuchen – von Metallkatalysatoren in Lösung bis zu photoaktiven Molekülen in biologischen Umgebungen – und dies mit deutlich weniger Trainingsdaten als zuvor.
Zitation: Barrett, R., Dietschreit, J.C.B. & Westermayr, J. Incorporating long-range interactions via the multipole expansion into ground and excited-state molecular simulations. npj Comput Mater 12, 135 (2026). https://doi.org/10.1038/s41524-026-02048-3
Schlüsselwörter: maschinell gelernte Potentiale, Quantenmechanik/Molekulmechanik, langreichweitige Elektrostatischen, molekulare Simulationen, angeregte Zustandsdynamik