Clear Sky Science · en
Incorporating long-range interactions via the multipole expansion into ground and excited-state molecular simulations
Why distant forces matter in chemistry
Many of the most important processes in chemistry and biology – from how drugs bind to proteins to how solar cells harvest light – depend on subtle electrical forces that act over surprisingly long distances. Accurately simulating these effects has traditionally required very expensive quantum mechanical calculations, which limits the size of systems and timescales that scientists can study. This paper introduces a new machine-learning approach, called Field-MACE, that makes it much more efficient to include these long-range forces when simulating molecules in complex environments such as liquids and biological matter.
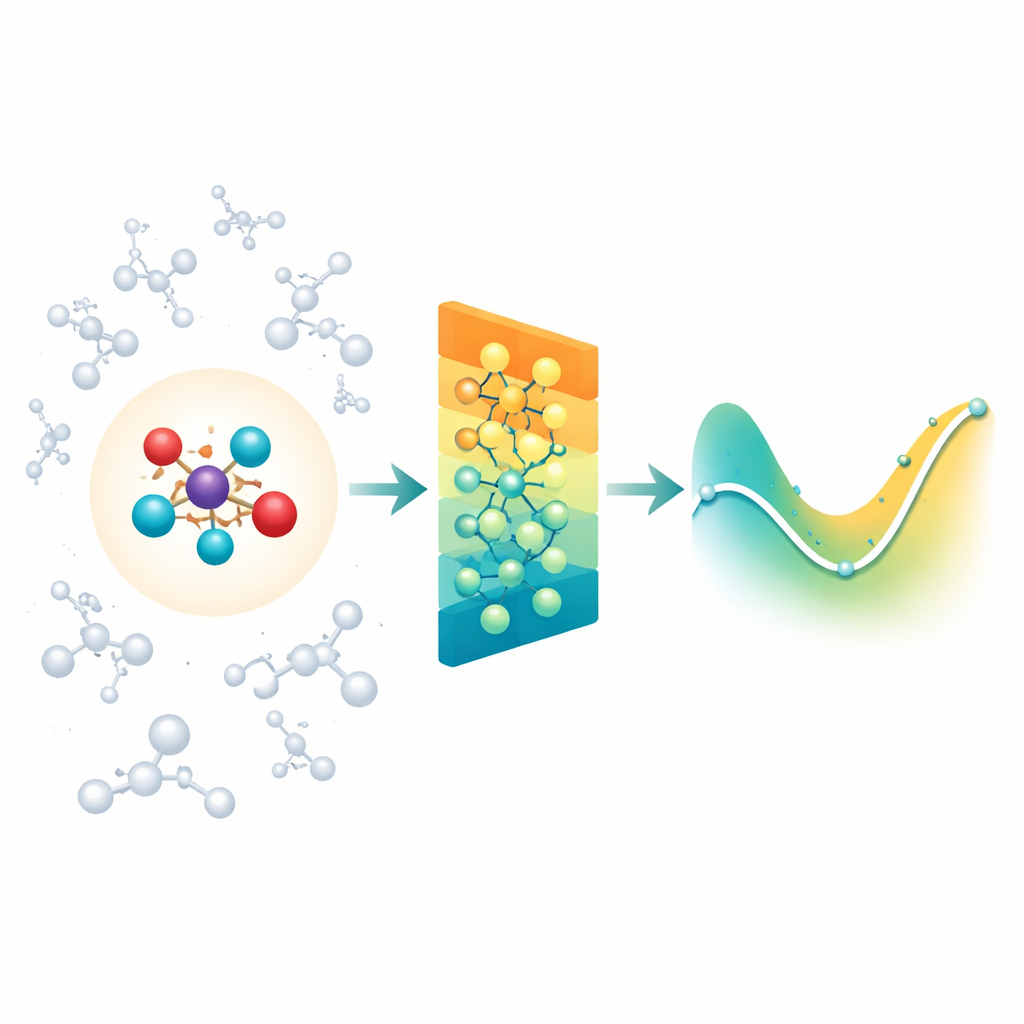
Blending detailed chemistry with its surroundings
When chemists simulate reactions in solution or inside proteins, they often use a “zoomed-in” quantum region where bonds are breaking and forming, surrounded by a larger “classical” region that provides the environment. The difficulty is that the environment still exerts long-range electrical forces on the reactive core, and most modern neural-network models for molecules mainly focus on interactions between close neighbors. As a result, they can miss important solvent or protein effects, or become impractically slow if they try to account for every distant interaction one by one. The authors tackle this by building on an existing class of rotation-aware neural networks (MACE) and extending them so that the quantum region can efficiently feel the influence of a large cloud of classical charges around it.
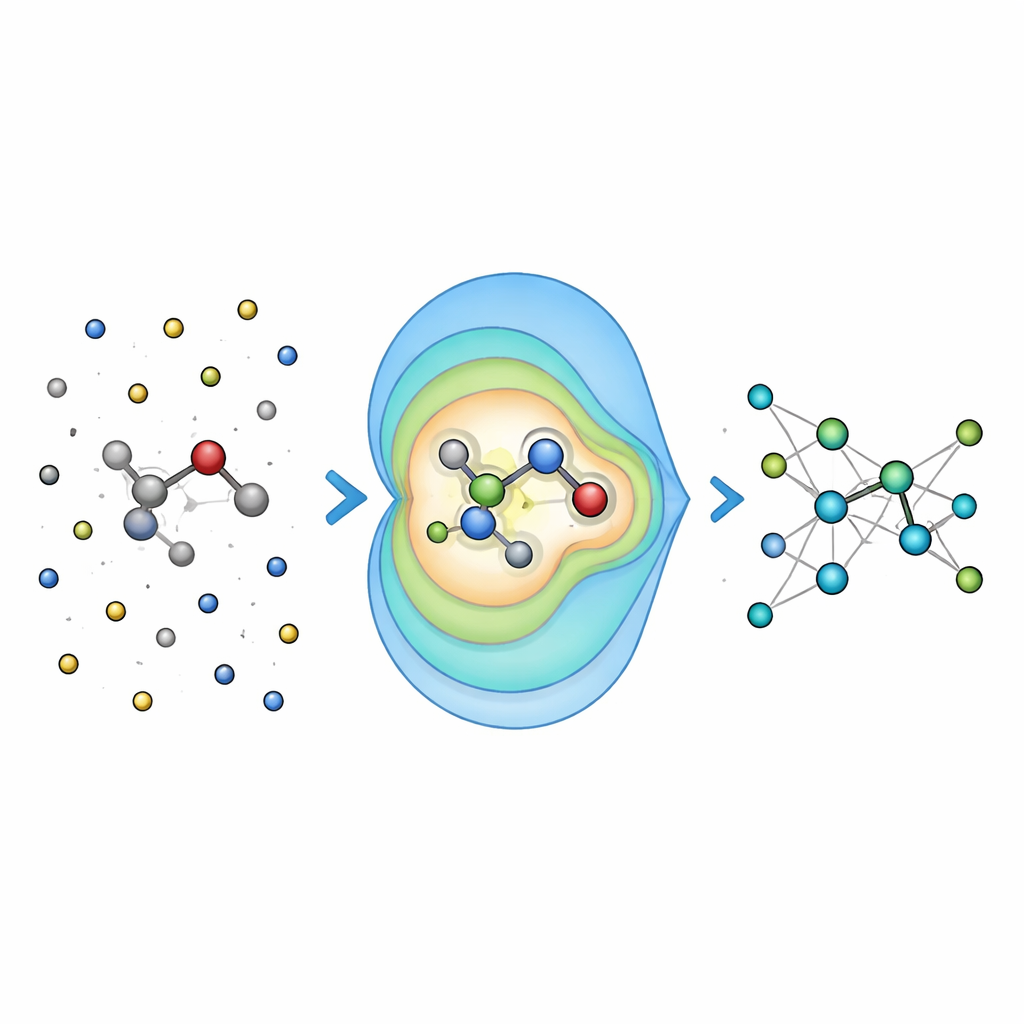
Summarizing distant charges into simple patterns
Instead of feeding the network every single interaction between the environment and each atom, Field-MACE uses a classic physics trick known as a multipole expansion. In simple terms, this compresses the influence of many charges into a small set of directional patterns: overall charge, average orientation of the electric field, and more refined shapes that describe how the field curves in space. These patterns are naturally expressed as angular shapes around the molecule and dovetail with how the underlying neural network represents geometry. The model combines two streams of information during learning: local, short-range messages that describe bonding and nearby neighbors, and long-range messages built from these multipole patterns that summarize how the surrounding solvent or material is tugging on the quantum region.
Testing accuracy on real-world chemical problems
To see whether this idea pays off, the researchers first benchmarked Field-MACE on molecules dissolved in liquids, comparing it to more traditional ways of handling long-range electrostatics, such as Ewald summation and simple Coulomb potentials. They found that including a modest number of multipole terms dramatically improved the accuracy of predicted energies and forces, especially for larger and more flexible molecules whose shapes are strongly influenced by their solvent. Crucially, this accuracy boost came with only a small increase in computational cost, because the expensive part of the calculation still scales mainly with the size of the quantum core, not with the sea of surrounding atoms.
From metal catalysts to light-driven reactions
The team then applied Field-MACE to two demanding case studies. In the first, they modeled nickel-based catalysts in liquid benzene, a class of metal complexes important in many industrial reactions. The new model produced stable molecular dynamics over many picoseconds and reproduced key structural features, such as bond lengths and angles, even for complexes that were not explicitly included in training. In the second case, they looked at a photochemical “ring-opening” reaction of the small molecule furan in water, a process that involves excited electronic states and rapid hops between them. Despite the complexity of these light-driven dynamics, Field-MACE closely matched high-level quantum simulations in how the population of different electronic states evolved over time.
Learning faster by reusing chemical knowledge
A major practical hurdle in such simulations is the scarcity and cost of reference data that already include environmental effects. The authors showed that they can dramatically cut this cost by starting from a pre-trained foundation model that was originally trained only on isolated molecules without solvent. By fine-tuning this existing "short-range" knowledge and adding the multipole-based long-range layer, Field-MACE reached high accuracy with far fewer data points. This was particularly striking for excited-state dynamics of furan: when only a few dozen expensive reference calculations were available, models trained from scratch failed, while those initialized from the foundation model still captured the essential reaction pathway.
What this means for future simulations
In everyday terms, Field-MACE gives chemists and materials scientists a way to keep the detailed quantum view where it matters while still feeling the pull of a large, complex environment – all without paying the usual computational price. By compressing distant electrical effects into a compact set of patterns and combining them with powerful neural networks, the method enables accurate, scalable simulations for both ground and excited states. This opens the door to studying more realistic systems – from metal catalysts in solution to photoactive molecules in biological settings – and to doing so with far less training data than before.
Citation: Barrett, R., Dietschreit, J.C.B. & Westermayr, J. Incorporating long-range interactions via the multipole expansion into ground and excited-state molecular simulations. npj Comput Mater 12, 135 (2026). https://doi.org/10.1038/s41524-026-02048-3
Keywords: machine learning potentials, quantum mechanics/molecular mechanics, long-range electrostatics, molecular simulations, excited-state dynamics