Clear Sky Science · es
Incorporación de interacciones de largo alcance mediante la expansión multipolar en simulaciones moleculares de estados fundamentales y excitados
Por qué importan las fuerzas a distancia en química
Muchos de los procesos más importantes en química y biología —desde cómo los fármacos se unen a proteínas hasta cómo las células solares capturan la luz— dependen de fuerzas eléctricas sutiles que actúan a distancias sorprendentemente largas. Simular con precisión estos efectos ha requerido tradicionalmente cálculos cuánticos muy costosos, lo que limita el tamaño de los sistemas y las escalas temporales que los científicos pueden estudiar. Este artículo presenta un nuevo enfoque de aprendizaje automático, llamado Field-MACE, que hace mucho más eficiente la inclusión de estas fuerzas de largo alcance al simular moléculas en entornos complejos como líquidos y materia biológica.
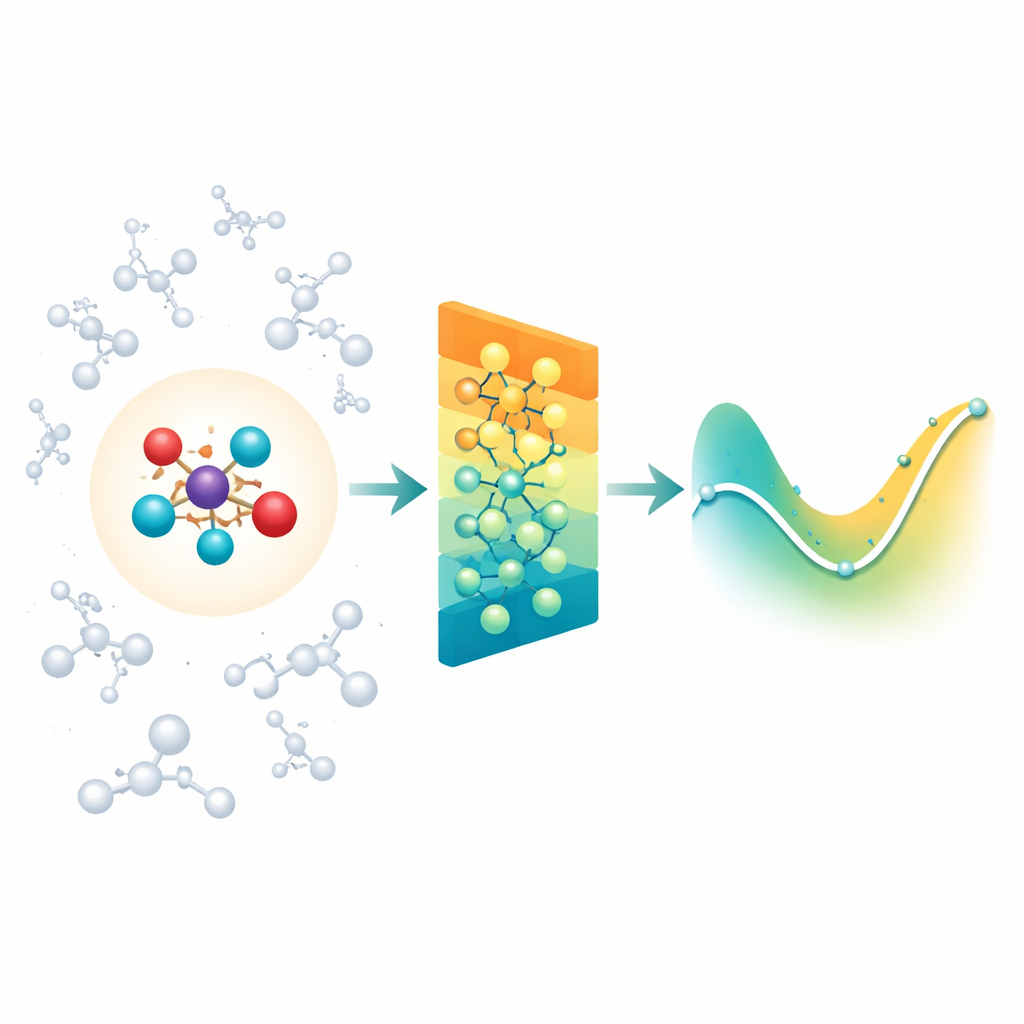
Combinar la química detallada con su entorno
Cuando los químicos simulan reacciones en disolución o dentro de proteínas, a menudo usan una región cuántica “ampliada” donde se rompen y forman enlaces, rodeada por una región “clásica” mayor que aporta el entorno. La dificultad es que el entorno sigue ejerciendo fuerzas eléctricas de largo alcance sobre el núcleo reactivo, y la mayoría de los modelos neuronales modernos para moléculas se centran principalmente en interacciones entre vecinos cercanos. Como resultado, pueden pasar por alto efectos importantes del disolvente o la proteína, o volverse imprácticamente lentos si intentan considerar cada interacción lejana una por una. Los autores abordan esto apoyándose en una clase existente de redes neuronales sensibles a la rotación (MACE) y extendiéndolas para que la región cuántica pueda sentir de manera eficiente la influencia de una gran nube de cargas clásicas a su alrededor.
Resumir cargas distantes en patrones simples
En lugar de alimentar a la red con cada interacción entre el entorno y cada átomo, Field-MACE utiliza un truco clásico de la física conocido como expansión multipolar. En términos sencillos, esto comprime la influencia de muchas cargas en un pequeño conjunto de patrones direccionales: carga total, orientación media del campo eléctrico y formas más refinadas que describen cómo el campo curva en el espacio. Estos patrones se expresan de forma natural como distribuciones angulares alrededor de la molécula y encajan con la manera en que la red neuronal subyacente representa la geometría. El modelo combina dos corrientes de información durante el aprendizaje: mensajes locales de corto alcance que describen enlaces y vecinos próximos, y mensajes de largo alcance construidos a partir de estos patrones multipolares que resumen cómo el disolvente o el material circundante tira de la región cuántica.
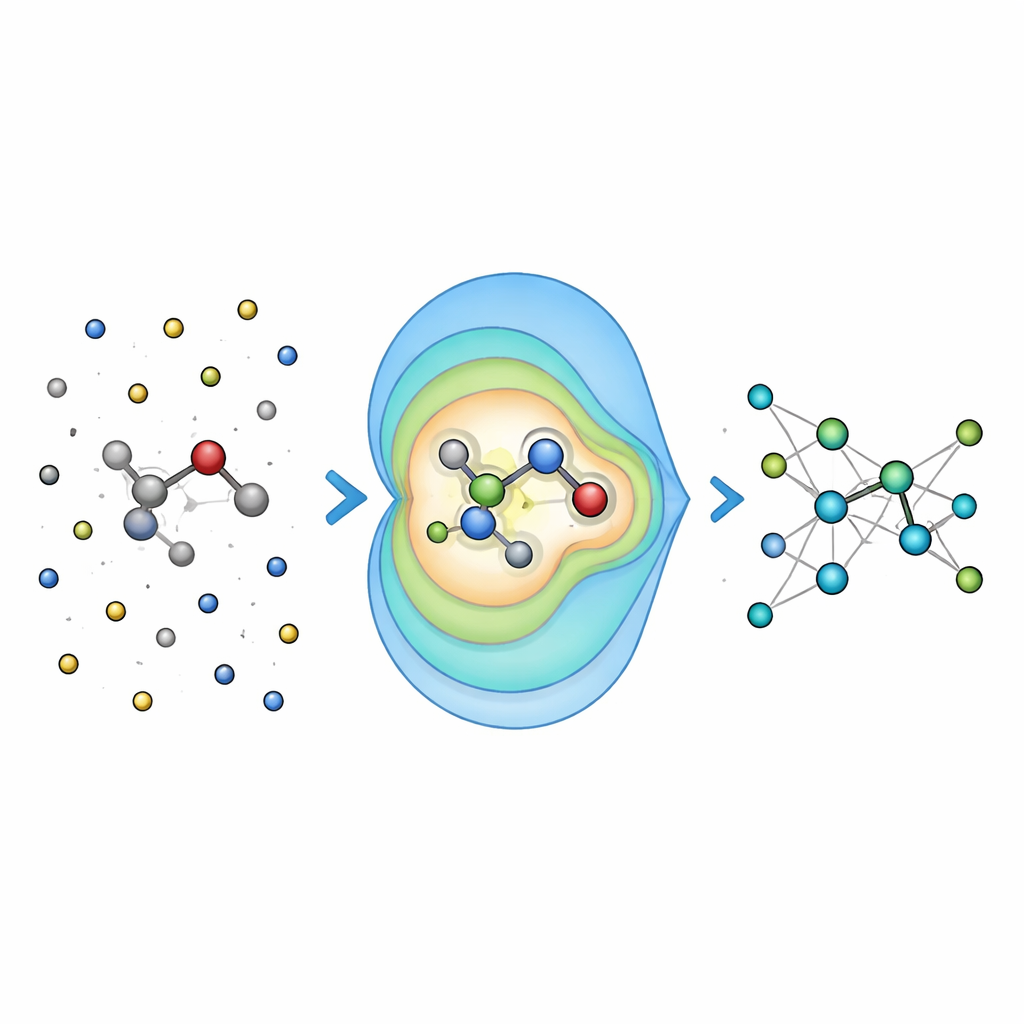
Probar la precisión en problemas químicos del mundo real
Para ver si la idea compensaba, los investigadores primero evaluaron Field-MACE en moléculas disueltas en líquidos, comparándolo con formas más tradicionales de manejar la electrostática de largo alcance, como la sumación de Ewald y los potenciales de Coulomb sencillos. Encontraron que incluir un número modesto de términos multipolares mejoró de forma drástica la precisión de las energías y fuerzas predichas, especialmente para moléculas más grandes y flexibles cuyas formas están fuertemente influenciadas por el disolvente. Crucialmente, este aumento de precisión supuso solo un pequeño incremento en el coste computacional, porque la parte cara del cálculo sigue escalando principalmente con el tamaño del núcleo cuántico, no con el mar de átomos circundantes.
Desde catalizadores metálicos hasta reacciones impulsadas por la luz
El equipo aplicó luego Field-MACE a dos estudios de caso exigentes. En el primero, modelaron catalizadores a base de níquel en benceno líquido, una clase de complejos metálicos importantes en muchas reacciones industriales. El nuevo modelo produjo dinámica molecular estable durante muchos picosegundos y reprodujo características estructurales clave, como longitudes y ángulos de enlace, incluso para complejos que no se incluyeron explícitamente en el entrenamiento. En el segundo caso, estudiaron una reacción fotoquímica de “apertura de anillo” del pequeño furan en agua, un proceso que implica estados electrónicos excitados y saltos rápidos entre ellos. A pesar de la complejidad de estas dinámicas impulsadas por la luz, Field-MACE se ajustó estrechamente a simulaciones cuánticas de alto nivel en cómo evolucionaba la población de los distintos estados electrónicos a lo largo del tiempo.
Aprender más rápido reutilizando conocimiento químico
Un obstáculo práctico importante en este tipo de simulaciones es la escasez y el coste de los datos de referencia que ya incluyen efectos ambientales. Los autores demostraron que pueden reducir drásticamente ese coste empezando desde un modelo base preentrenado que originalmente se había entrenado solo con moléculas aisladas sin disolvente. Al ajustar finamente este conocimiento “de corto alcance” existente y añadir la capa de largo alcance basada en multipolos, Field-MACE alcanzó alta precisión con muchos menos puntos de datos. Esto fue particularmente notable para la dinámica de estados excitados del furan: cuando solo había disponibles unas pocas docenas de cálculos de referencia costosos, los modelos entrenados desde cero fracasaron, mientras que los inicializados desde el modelo base aún capturaron la vía de reacción esencial.
Qué significa esto para futuras simulaciones
En términos cotidianos, Field-MACE ofrece a químicos y científicos de materiales una forma de mantener la vista cuántica detallada donde importa al tiempo que sienten el tirón de un entorno grande y complejo —todo ello sin pagar el habitual precio computacional. Al comprimir los efectos eléctricos lejanos en un conjunto compacto de patrones y combinarlos con redes neuronales potentes, el método permite simulaciones precisas y escalables tanto para estados fundamentales como excitados. Esto abre la puerta a estudiar sistemas más realistas —desde catalizadores metálicos en disolución hasta moléculas fotoactivas en entornos biológicos— y a hacerlo con muchos menos datos de entrenamiento que antes.
Cita: Barrett, R., Dietschreit, J.C.B. & Westermayr, J. Incorporating long-range interactions via the multipole expansion into ground and excited-state molecular simulations. npj Comput Mater 12, 135 (2026). https://doi.org/10.1038/s41524-026-02048-3
Palabras clave: potenciales de aprendizaje automático, mecánica cuántica/mecánica molecular, electrostática de largo alcance, simulaciones moleculares, dinámica de estados excitados