Clear Sky Science · pt
Incorporação de interações de longo alcance via expansão multipolar em simulações moleculares de estados fundamental e excitado
Por que forças distantes importam na química
Muitos dos processos mais importantes na química e na biologia — desde como fármacos se ligam a proteínas até como células solares capturam luz — dependem de forças elétricas sutis que atuam a distâncias surpreendentemente grandes. Simular esses efeitos com precisão tradicionalmente exige cálculos quânticos muito caros, o que limita o tamanho dos sistemas e as escalas de tempo que os pesquisadores podem estudar. Este artigo apresenta uma nova abordagem por aprendizado de máquina, chamada Field-MACE, que torna muito mais eficiente a inclusão dessas forças de longo alcance ao simular moléculas em ambientes complexos, como líquidos e matéria biológica.
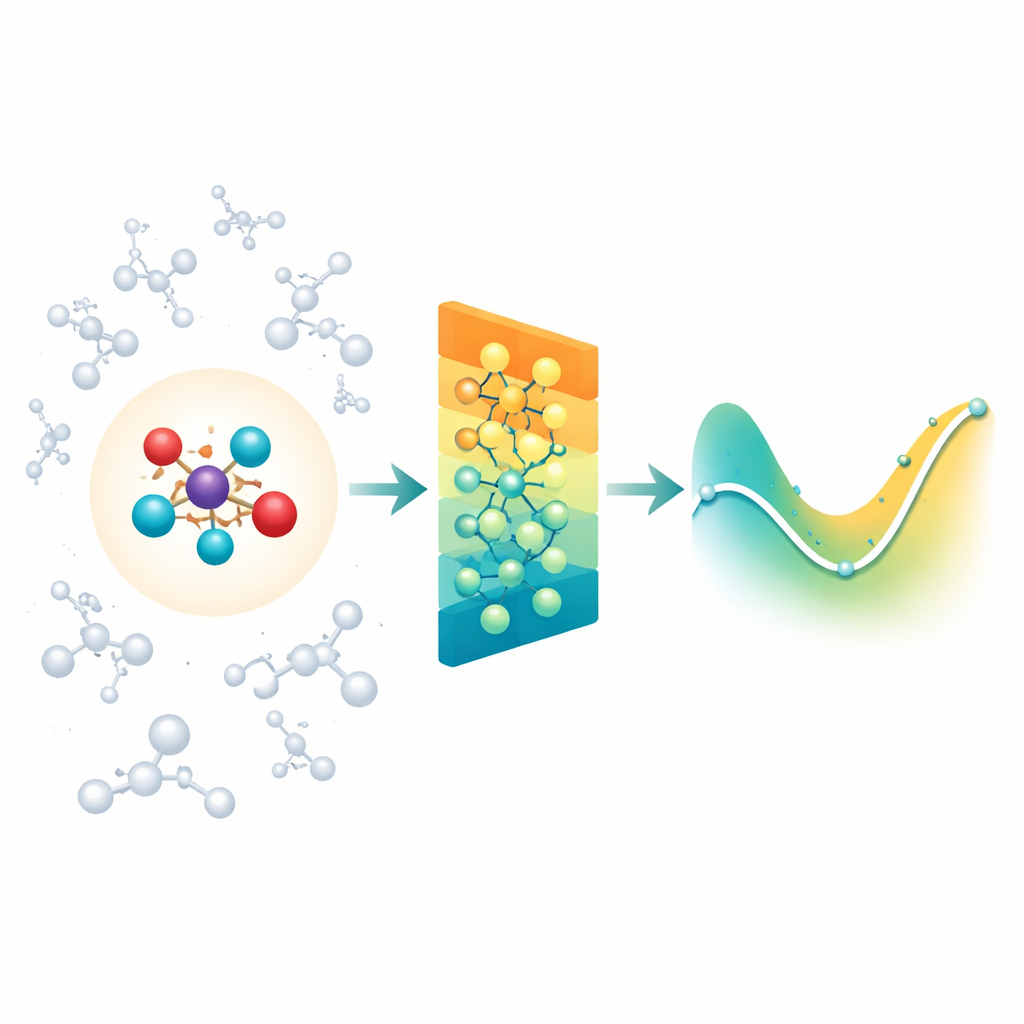
Mesclando a química detalhada com seu entorno
Quando químicos simulam reações em solução ou no interior de proteínas, costumam usar uma região quântica “ampliada” onde ligações se rompem e se formam, rodeada por uma região “clássica” maior que fornece o ambiente. A dificuldade é que o entorno ainda exerce forças elétricas de longo alcance sobre o núcleo reativo, e a maioria dos modelos modernos de redes neurais para moléculas foca principalmente em interações entre vizinhos próximos. Como resultado, eles podem deixar de capturar efeitos importantes do solvente ou da proteína, ou tornar-se impraticavelmente lentos se tentarem contabilizar cada interação distante uma por uma. Os autores enfrentam isso ao ampliar uma classe existente de redes neurais sensíveis à rotação (MACE) para que a região quântica possa sentir eficientemente a influência de uma grande nuvem de cargas clássicas ao seu redor.
Resumindo cargas distantes em padrões simples
Em vez de alimentar a rede com cada interação entre o ambiente e cada átomo, o Field-MACE usa um truque clássico da física conhecido como expansão multipolar. Em termos simples, isso comprime a influência de muitas cargas em um pequeno conjunto de padrões direcionais: carga total, orientação média do campo elétrico e formas mais refinadas que descrevem como o campo curva no espaço. Esses padrões são naturalmente expressos como formas angulares ao redor da molécula e se encaixam com a maneira como a rede neural subjacente representa a geometria. O modelo combina dois fluxos de informação durante o aprendizado: mensagens locais de curto alcance que descrevem ligações e vizinhos próximos, e mensagens de longo alcance construídas a partir desses padrões multipolares que resumem como o solvente ou material circundante puxa a região quântica.
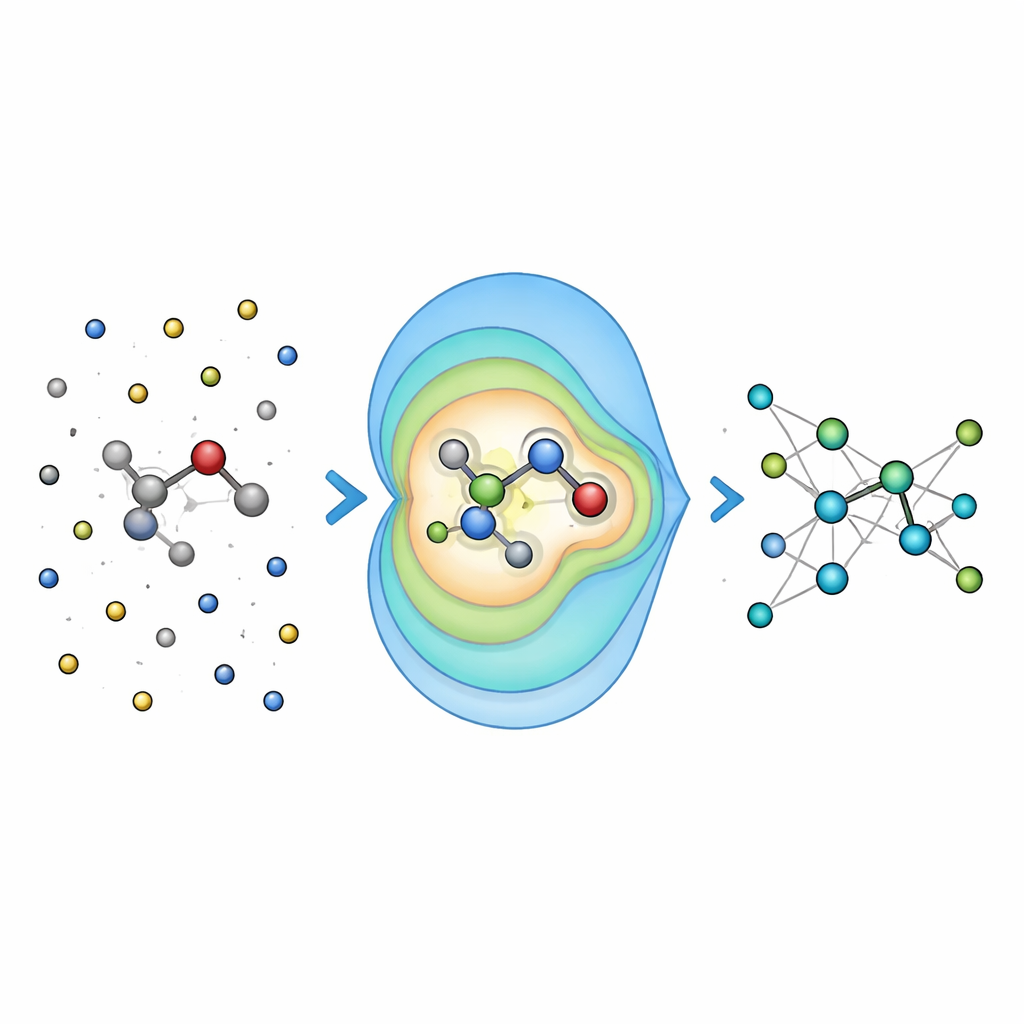
Testando a precisão em problemas químicos do mundo real
Para verificar se a ideia compensa, os pesquisadores primeiro avaliaram o Field-MACE em moléculas dissolvidas em líquidos, comparando-o com maneiras mais tradicionais de lidar com eletrostática de longo alcance, como soma de Ewald e potenciais de Coulomb simples. Eles descobriram que incluir um número moderado de termos multipolares melhorou dramaticamente a precisão das energias e forças previstas, especialmente para moléculas maiores e mais flexíveis cujas formas são fortemente influenciadas pelo solvente. Crucialmente, esse ganho de precisão veio com apenas um pequeno aumento no custo computacional, porque a parte cara do cálculo ainda escala principalmente com o tamanho do núcleo quântico, e não com o mar de átomos circundantes.
De catalisadores metálicos a reações induzidas por luz
A equipe então aplicou o Field-MACE a dois estudos de caso exigentes. No primeiro, modelaram catalisadores à base de níquel em benzeno líquido, uma classe de complexos metálicos importante em muitas reações industriais. O novo modelo produziu dinâmica molecular estável por muitos picosegundos e reproduziu características estruturais chave, como comprimentos e ângulos de ligação, mesmo para complexos que não haviam sido incluídos explicitamente no treinamento. No segundo caso, observaram uma reação fotoquímica de “abertura de anel” da pequena molécula furano em água, um processo que envolve estados eletrônicos excitados e saltos rápidos entre eles. Apesar da complexidade dessa dinâmica induzida pela luz, o Field-MACE combinou de perto com simulações quânticas de alto nível em como a população de diferentes estados eletrônicos evoluiu ao longo do tempo.
Aprendendo mais rápido ao reaproveitar conhecimento químico
Um obstáculo prático importante nessas simulações é a escassez e o custo de dados de referência que já incluam efeitos ambientais. Os autores mostraram que podem reduzir dramaticamente esse custo ao partir de um modelo fundacional pré-treinado que originalmente foi treinado apenas em moléculas isoladas sem solvente. Ao ajustar esse conhecimento “de curto alcance” existente e adicionar a camada de longo alcance baseada em multipolos, o Field-MACE alcançou alta precisão com muito menos pontos de dados. Isso foi particularmente marcante para a dinâmica de estados excitados do furano: quando havia apenas algumas dezenas de cálculos de referência caros disponíveis, modelos treinados do zero falharam, enquanto aqueles inicializados a partir do modelo fundacional ainda capturaram a via reacional essencial.
O que isso significa para simulações futuras
Em termos práticos, o Field-MACE dá a químicos e cientistas de materiais uma maneira de manter a visão quântica detalhada onde ela importa, sentindo ao mesmo tempo a influência de um ambiente grande e complexo — tudo isso sem pagar o custo computacional habitual. Ao comprimir efeitos elétricos distantes em um conjunto compacto de padrões e combiná-los com redes neurais poderosas, o método possibilita simulações precisas e escaláveis tanto para estados fundamentais quanto excitados. Isso abre a porta para estudar sistemas mais realistas — de catalisadores metálicos em solução a moléculas fotoativas em ambientes biológicos — e fazê-lo com muito menos dados de treinamento do que antes.
Citação: Barrett, R., Dietschreit, J.C.B. & Westermayr, J. Incorporating long-range interactions via the multipole expansion into ground and excited-state molecular simulations. npj Comput Mater 12, 135 (2026). https://doi.org/10.1038/s41524-026-02048-3
Palavras-chave: potenciais por aprendizado de máquina, mecânica quântica/mecânica molecular, eletrostática de longo alcance, simulações moleculares, dinâmica de estados excitados