Clear Sky Science · fr
Intégrer les interactions à longue portée via le développement multipolaire dans les simulations moléculaires des états fondamentaux et excités
Pourquoi les forces lointaines comptent en chimie
Beaucoup des processus les plus importants en chimie et en biologie – depuis la liaison des médicaments aux protéines jusqu’à la capture de la lumière par les cellules solaires – dépendent de forces électriques subtiles qui s’exercent sur des distances étonnamment longues. Simuler ces effets avec précision a traditionnellement exigé des calculs de mécanique quantique très coûteux, ce qui limite la taille des systèmes et les échelles de temps accessibles aux chercheurs. Cet article présente une nouvelle approche par apprentissage automatique, appelée Field-MACE, qui rend beaucoup plus efficace l’incorporation de ces forces à longue portée lors de la simulation de molécules dans des environnements complexes tels que des liquides et des milieux biologiques.
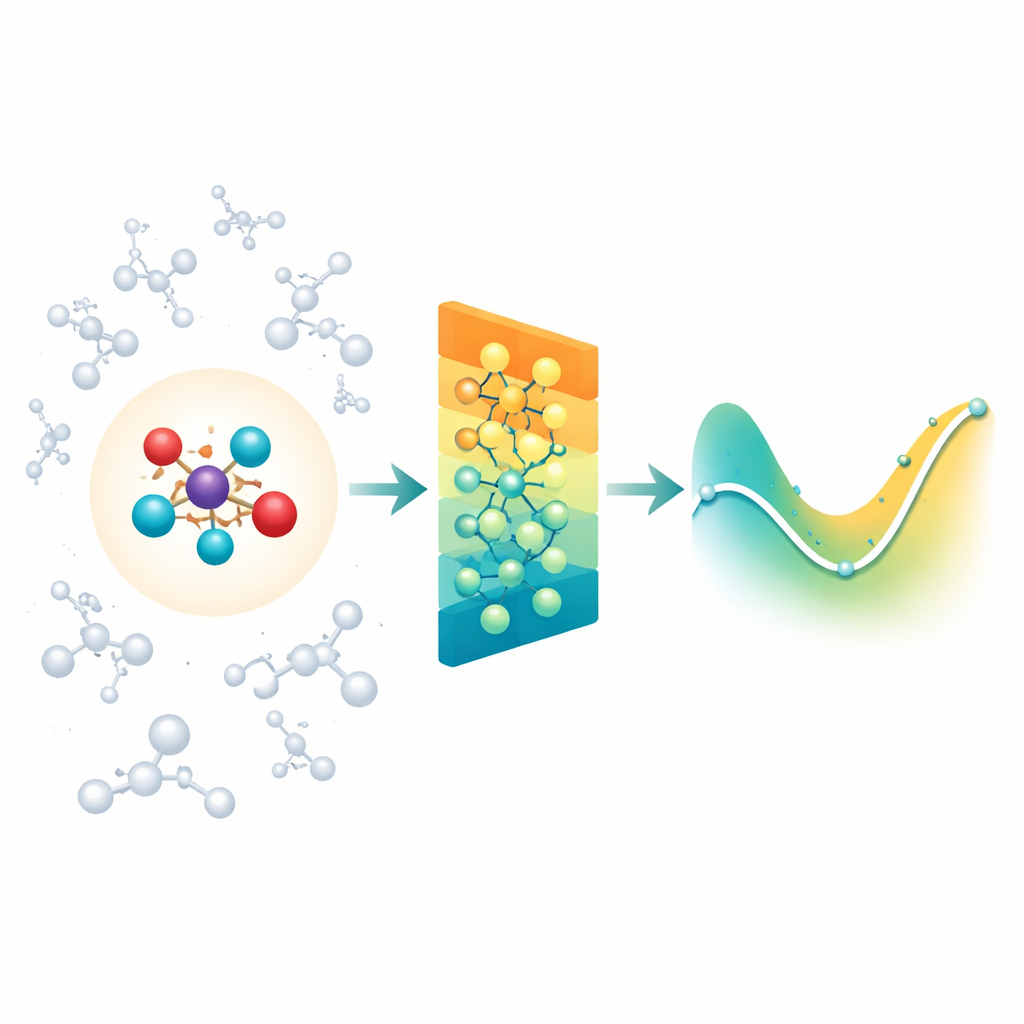
Allier la chimie détaillée à son environnement
Lorsque les chimistes simulent des réactions en solution ou à l’intérieur de protéines, ils utilisent souvent une région quantique « zoomée » où les liaisons se brisent et se forment, entourée d’une plus grande région « classique » qui constitue l’environnement. Le problème est que cet environnement exerce néanmoins des forces électriques à longue portée sur le cœur réactif, et la plupart des modèles neuronaux modernes pour molécules se concentrent surtout sur les interactions entre voisins proches. Ils peuvent donc manquer des effets importants du solvant ou de la protéine, ou devenir impraticables si l’on tente de prendre en compte chaque interaction lointaine une à une. Les auteurs s’attaquent à cette difficulté en s’appuyant sur une classe existante de réseaux neuronaux sensibles aux rotations (MACE) et en les étendant pour que la région quantique puisse ressentir efficacement l’influence d’un large nuage de charges classiques qui l’entoure.
Résumer les charges lointaines en motifs simples
Plutôt que d’alimenter le réseau avec chaque interaction entre l’environnement et chaque atome, Field-MACE utilise un procédé classique de la physique connu sous le nom de développement multipolaire. En termes simples, cela compresse l’influence de nombreuses charges en un petit ensemble de motifs directionnels : charge globale, orientation moyenne du champ électrique, et formes plus raffinées décrivant la façon dont le champ se courbe dans l’espace. Ces motifs s’expriment naturellement comme des formes angulaires autour de la molécule et s’accordent avec la manière dont le réseau neuronal sous-jacent représente la géométrie. Le modèle combine deux flux d’information pendant l’apprentissage : des messages locaux à courte portée qui décrivent les liaisons et les voisins proches, et des messages à longue portée construits à partir de ces motifs multipolaires qui résument la façon dont le solvant ou le matériau environnant attire la région quantique.
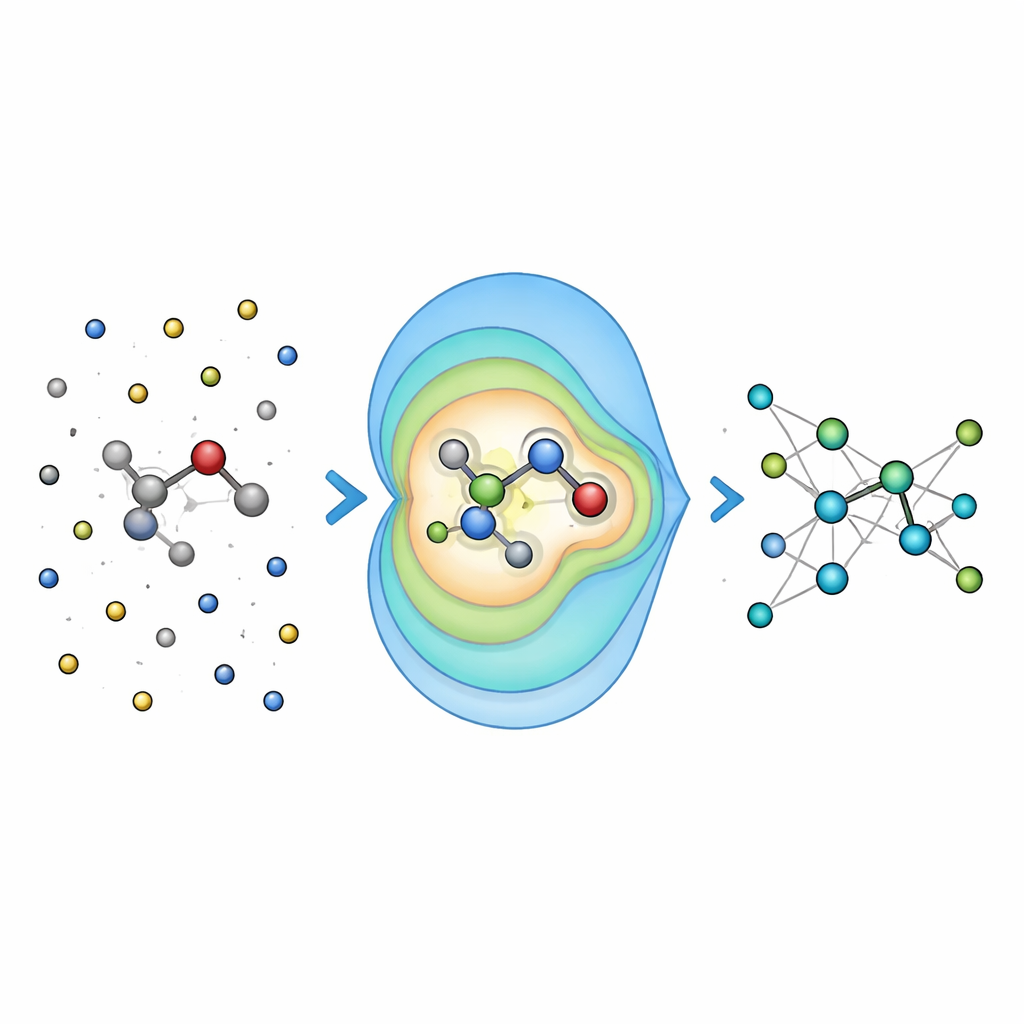
Tester la précision sur des problèmes chimiques réels
Pour vérifier si cette idée est payante, les chercheurs ont d’abord évalué Field-MACE sur des molécules dissoutes dans des liquides, en le comparant à des méthodes plus traditionnelles de prise en compte de l’électrostatique à longue portée, comme la sommation d’Ewald et les potentiels coulombiens simples. Ils ont constaté que l’inclusion d’un nombre modeste de termes multipolaires améliorait considérablement la précision des énergies et des forces prédites, en particulier pour les molécules plus grandes et plus flexibles dont la forme est fortement influencée par le solvant. De manière cruciale, ce gain de précision s’accompagnait d’une augmentation négligeable du coût de calcul, car la partie coûteuse du calcul reste principalement fonction de la taille du noyau quantique, et non de la mer d’atomes environnante.
Des catalyseurs métalliques aux réactions induites par la lumière
L’équipe a ensuite appliqué Field-MACE à deux études de cas exigeantes. Dans la première, ils ont modélisé des catalyseurs à base de nickel en benzène liquide, une classe de complexes métalliques importants dans de nombreuses réactions industrielles. Le nouveau modèle a produit une dynamique moléculaire stable sur plusieurs picosecondes et a reproduit des caractéristiques structurales clés, telles que les longueurs et angles de liaison, même pour des complexes qui n’avaient pas été inclus explicitement dans l’entraînement. Dans le second cas, ils ont examiné une réaction photochimique d’« ouverture de cycle » de la petite molécule furane en eau, un processus impliquant des états électroniques excités et des sauts rapides entre eux. Malgré la complexité de ces dynamiques induites par la lumière, Field-MACE a très bien reproduit les simulations quantiques de haut niveau quant à l’évolution temporelle des populations des différents états électroniques.
Apprendre plus vite en réutilisant des connaissances chimiques
Un obstacle pratique majeur dans ce type de simulations est la rareté et le coût des données de référence qui incluent déjà les effets environnementaux. Les auteurs montrent qu’il est possible de réduire considérablement ce coût en partant d’un modèle de base pré-entraîné, initialement entraîné uniquement sur des molécules isolées sans solvant. En ajustant finement ces connaissances « à courte portée » existantes et en ajoutant la couche longue portée basée sur les multipôles, Field-MACE a atteint une grande précision avec beaucoup moins de points de données. Cela a été particulièrement frappant pour la dynamique des états excités du furane : lorsque seules quelques dizaines de calculs de référence coûteux étaient disponibles, les modèles entraînés à partir de zéro échouaient, tandis que ceux initialisés à partir du modèle de base capturaient encore la voie réactionnelle essentielle.
Ce que cela implique pour les simulations futures
En termes simples, Field-MACE offre aux chimistes et aux scientifiques des matériaux un moyen de conserver la vue quantique détaillée là où elle compte tout en ressentant l’influence d’un environnement large et complexe – et ce sans payer le coût de calcul habituel. En compressant les effets électriques lointains en un ensemble compact de motifs et en les combinant avec de puissants réseaux neuronaux, la méthode permet des simulations précises et évolutives pour les états fondamentaux comme pour les états excités. Cela ouvre la voie à l’étude de systèmes plus réalistes – depuis des catalyseurs métalliques en solution jusqu’à des molécules photoactives en milieu biologique – et à le faire avec bien moins de données d’entraînement qu’auparavant.
Citation: Barrett, R., Dietschreit, J.C.B. & Westermayr, J. Incorporating long-range interactions via the multipole expansion into ground and excited-state molecular simulations. npj Comput Mater 12, 135 (2026). https://doi.org/10.1038/s41524-026-02048-3
Mots-clés: potentiels par apprentissage automatique, mécanique quantique/mécanique moléculaire, électrostatique à longue portée, simulations moléculaires, dynamique des états excités