Clear Sky Science · sv
Systemberoende reparametrisering av SCAN‑funktionalen för korrekta bandgap: från analytiska begränsningar till maskininlärning
Varför justering av bandgap spelar roll
Från smartphones och solpaneler till lysdioder och sensorer bygger många moderna teknologier på halvledare — material vars förmåga att leda elektricitet bestäms av en nyckelstorhet kallad bandgap. Detta bandgap är beryktat svårt att beräkna exakt på en dator utan att använda kostsamma metoder. Artikeln undersöker ett smartare, mer flexibelt sätt att finjustera ett befintligt, allmänt använt kvantmekaniskt recept så att det kan förutsäga bandgap för många fasta ämnen mer tillförlitligt, utan att betala den höga kostnaden för de mest avancerade teknikerna.
En smartare ratt på en betrodd teori
Författarna arbetar inom densitetsfunktionalteori, ett standardsätt att simulera material på atomär nivå. I denna ram är den mest känsliga ingrediensen utbytes–korrelations ”funktionen”, en formel som kodar hur elektroner interagerar. En populär version kallad SCAN bygger på rigorösa fysikaliska begränsningar snarare än omfattande passning till data, vilket gör den robust för många egenskaper men endast måttligt exakt för bandgap. Istället för att uppfinna en helt ny formel frågar författarna: vad händer om vi varsamt fininställer ett fåtal interna numeriska rattar i SCAN för varje material, så att den bättre speglar varje solids bindningsmiljö?
Justera receptet för olika typer av material
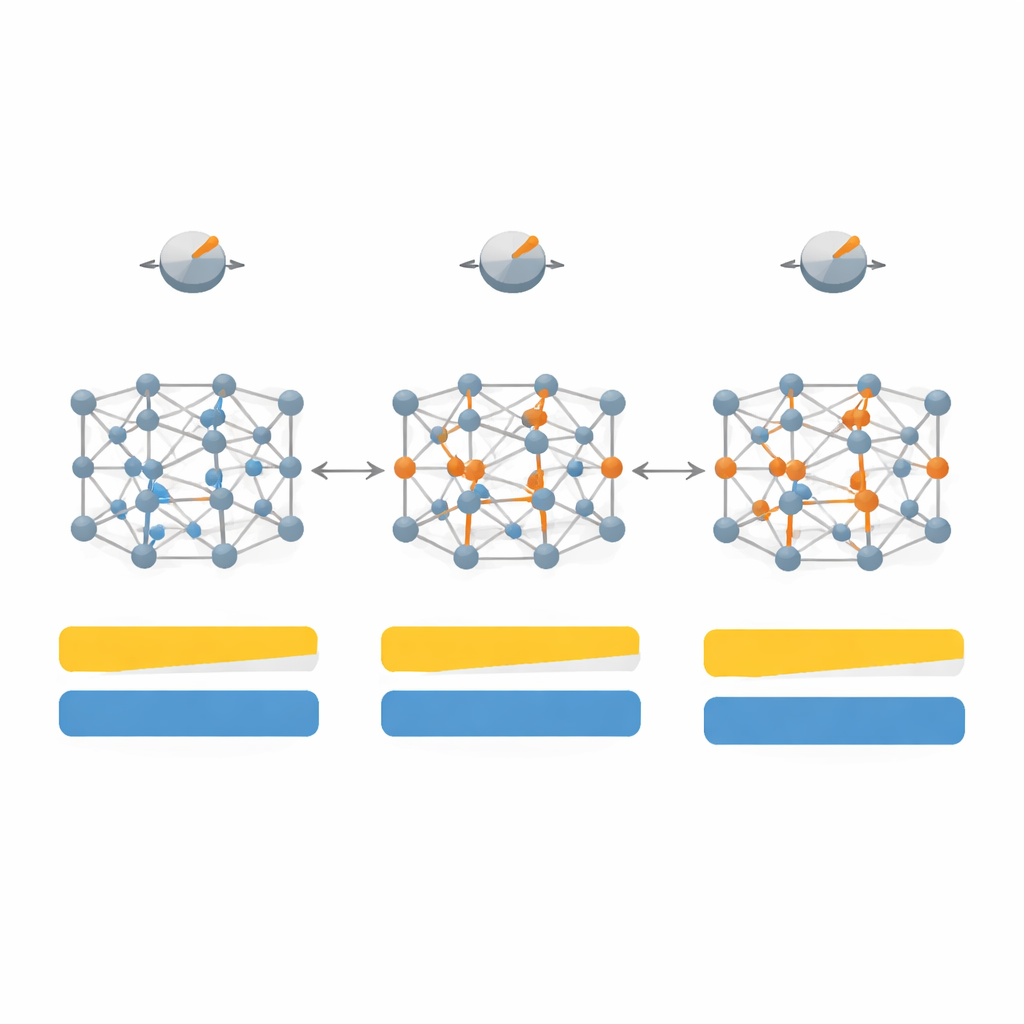
De kartlägger först hur förändringar i sju nyckelparametrar inom SCAN påverkar både bandgap och kristallstruktur för ett urval av 20 representativa material, inklusive klassiska halvledare som kisel och galliumarsenid, tröga ädelgaskristaller och starkt joniska salter såsom litiumfluorid. Genom att utforska detta högdimensionella parameterutrymme, och sedan begränsa det till de fyra mest inflytelserika rattarna, utformar de en ”systemberoende” version av funktionen som de kallar SD‑SCAN. För kovalenta material — där atomer delar elektroner i riktade bindningar — kan noggranna justeringar pressa de beräknade bandgapen nära experimentella värden samtidigt som gitterkonstanterna, kristallens grundläggande mått, i stort sett förblir korrekta. I kontrast förblir starkt joniska material, där elektroner ligger mer rigidt på en viss atomtyp, svåra att åtgärda med detta tillvägagångssätt, vilket avslöjar en djupare begränsning i den underliggande formeln.
Hur elektroner och ljus svarar
Utöver att bara matcha bandgapens storlek undersöker teamet hur deras omstämda funktional påverkar den detaljerade elektronstrukturen och hur elektroner fördelas mellan atomerna. För kovalenta kristaller som diamant och kisel förskjuter SD‑SCAN de tomma energibanden uppåt och sprider laddningen mer realistiskt längs bindningarna, vilket stärker deras delade‑elektronkaraktär. När de beräknar optiska egenskaper — hur materialet svarar på ljus och högfrekventa elektriska fält — leder de förbättrade bandgapen till mer korrekta dielektricitetskonstanter och bättre överensstämmelse med uppmätta spektra, särskilt för material där original‑SCAN antingen underskattade gapet eller till och med felaktigt förutsade metalliskt beteende.
Låta data välja rätt rattar
Att manuellt ställa parametrar för varje nytt material skulle snabbt bli orealistiskt, så författarna vänder sig till maskininlärning för att automatisera processen. De sammanställer en större dataset med 260 halvledare och isolatorer och bestämmer för var och en vilka parameterinställningar som fungerar bäst. Med enkla deskriptorer — som kristallsymmetri, hur starkt atomer delar eller överför elektroner, och hur tätt packade atomerna är — tränar de regressionsmodeller för att förutsäga SCAN:s optimala interna inställningar enbart utifrån dessa fysikaliska egenskaper. Deras bästa modell, en multipel linjär regression, kan uppskatta bandgap med betydligt lägre genomsnittligt fel än standard‑SCAN och närmar sig det ideala, fullständigt handstämda SD‑SCAN, samtidigt som den behåller i stort sett samma låga beräkningskostnad.
En genväg för vardagliga beräkningar
Som ett ännu enklare alternativ identifierar författarna en avgörande parameter som särskilt påverkar halvledare och isolatorer och föreslår en fast omstämning de kallar SCAN‑0.2. Denna variant bevarar all SCAN‑fysik men formar om hur olika bindningsmiljöer behandlas. Över deras stora testmängd förbättrar SCAN‑0.2 systematiskt bandgapförutsägelser jämfört med originalfunktionen samtidigt som mycket korrekta kristallstrukturer bibehålls. Den presterar i många system på samma nivå som maskininlärningsmetoden och erbjuder ett bekvämt drop‑in‑ersättningsalternativ för rutinmässig materialmodellering.
Vad detta betyder för framtida materialdesign
Enkelt uttryckt visar studien att ett vida använt teoretiskt verktyg för materialimulering kan göras märkbart bättre på att förutsäga bandgap genom att lära sig hur dess interna rattar ska ställas in beroende på vilken typ av material som studeras. För kovalenta halvledare — elektronikens arbetsdjur — ger metoden gap och optiska svar nära experiment till en bråkdel av kostnaden för de mest avancerade metoderna. Samtidigt understryker de kvarstående svårigheterna med starkt joniska föreningar var ny fysik, inte bara bättre passning, behövs. Tillsammans pekar den systemberoende SD‑SCAN, den datadrivna ML‑SCAN och den enkla SCAN‑0.2‑varianten mot en framtid där utbytes–korrelationsrecept inte längre är one‑size‑fits‑all utan adaptivt skräddarsys för varje material, vilket påskyndar datorstyrd upptäckt av bättre elektroniska och fotoniska material.
Citering: Dovale-Farelo, V., Tavadze, P., Marques, M.A.L. et al. System-conditioned reparameterization of the SCAN functional for accurate bandgaps: from analytical constraints to machine learning. npj Comput Mater 12, 162 (2026). https://doi.org/10.1038/s41524-026-02009-w
Nyckelord: halledarbandgap, densitetsfunktionalteori, SCAN‑funktionen, maskininlärning i materialvetenskap, elektronstruktur