Clear Sky Science · pl
Reparametryzacja funkcjonału SCAN zależna od układu dla dokładnych przerw energetycznych: od ograniczeń analitycznych po uczenie maszynowe
Dlaczego regulacja przerw energetycznych ma znaczenie
Od smartfonów i paneli słonecznych po diody LED i sensory — wiele współczesnych technologii opiera się na półprzewodnikach, których zdolność przewodzenia prądu kontroluje kluczowa wielkość zwana przerwą energetyczną. Obliczanie tej przerwy na komputerze jest znane jako trudne do wykonania z wysoką dokładnością bez użycia kosztownych metod. Artykuł bada sprytniejszy, bardziej elastyczny sposób dostrojenia istniejącego, powszechnie stosowanego przepisu kwantowo‑mechanicznego, tak aby mógł bardziej wiarygodnie przewidywać przerwy energetyczne wielu ciał stałych, bez ponoszenia wysokich kosztów najzaawansowanych technik.
Inteligentniejsze pokrętło w zaufanej teorii
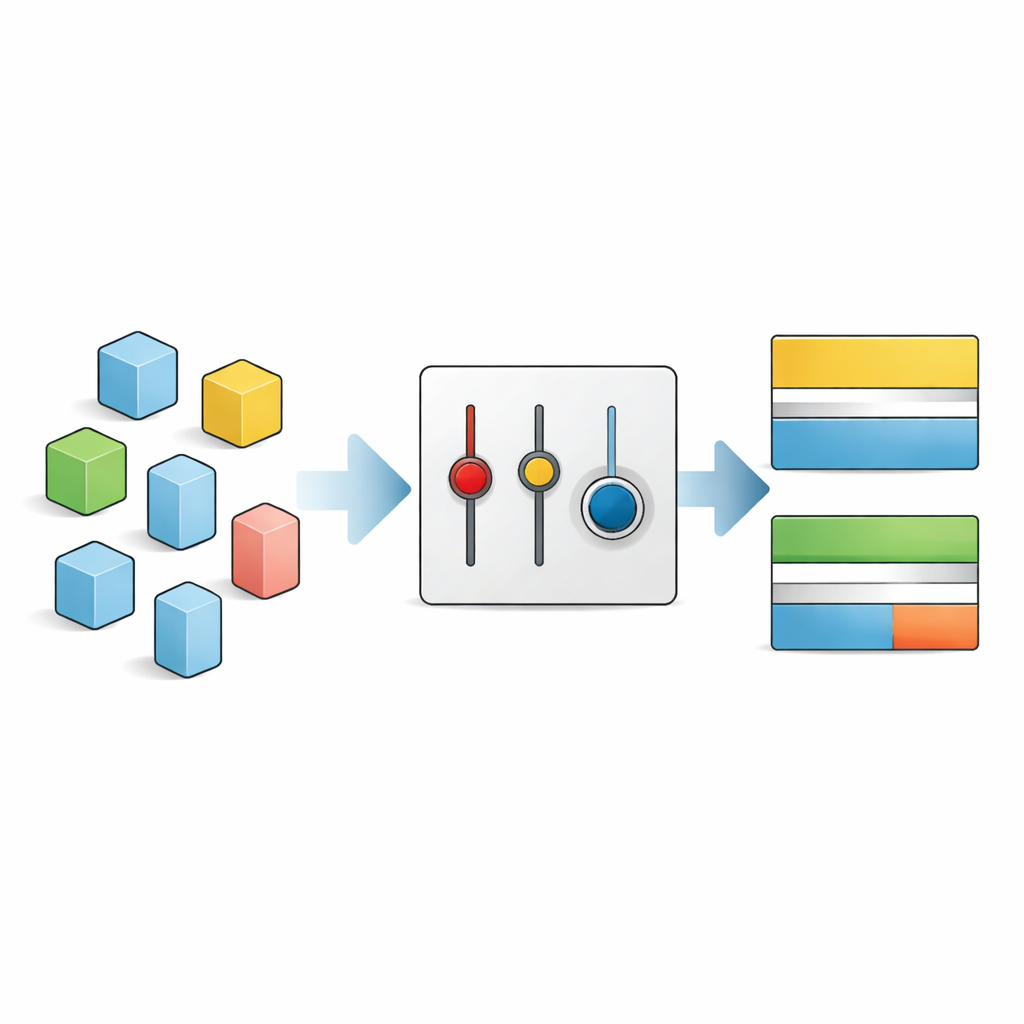
Autorzy pracują w ramach teorii funkcjonału gęstości, standardowego podejścia do symulacji materiałów na poziomie atomowym. W tym schemacie najdelikatniejszym składnikiem jest wymiana–korelacja „funkcjonał” — formuła opisująca, jak elektrony się oddziałują. Popularna wersja o nazwie SCAN jest zbudowana na rygorystycznych ograniczeniach fizycznych, a nie na intensywnym dopasowywaniu do danych, co czyni ją solidną dla wielu własności, ale tylko umiarkowanie dokładną dla przerw energetycznych. Zamiast wymyślać zupełnie nową formułę, autorzy pytają: co się stanie, jeśli delikatnie przestroimy kilka wewnętrznych numerycznych pokręteł w SCAN dla każdego materiału z osobna, aby lepiej odzwierciedlał środowisko wiązań w badanym ciele stałym?
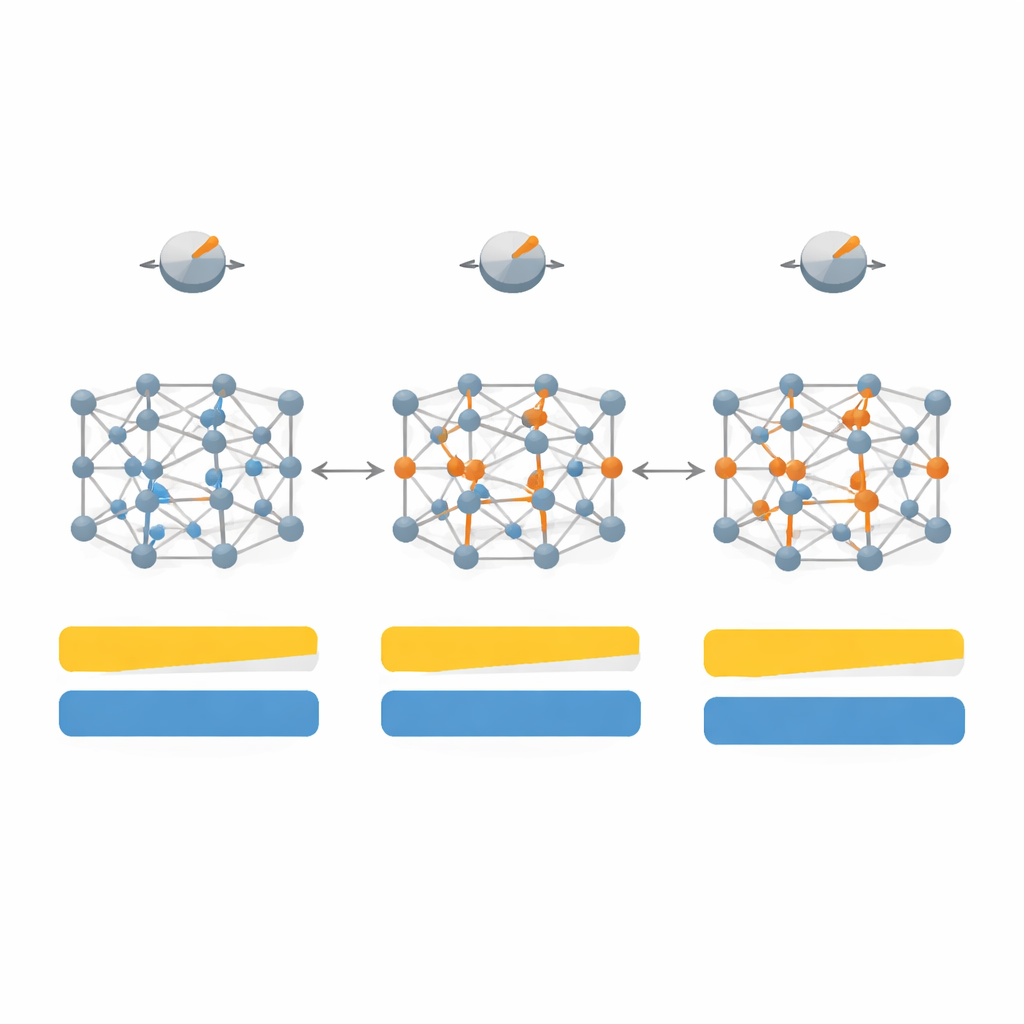
Dostrojenie przepisu dla różnych rodzajów ciał stałych
Pierwsze kroki polegały na zmapowaniu, jak zmiany siedmiu kluczowych parametrów w SCAN wpływają zarówno na przerwę energetyczną, jak i na strukturę krystaliczną dla zestawu 20 reprezentatywnych materiałów, w tym klasycznych półprzewodników jak krzem i arsenek galu, inercyjnych kryształów gazów szlachetnych oraz silnie jonowych soli, takich jak fluorek litu. Eksplorując tę wielowymiarową przestrzeń parametrów, a następnie zawężając ją do czterech najbardziej wpływowych pokręteł, zaprojektowali „zależną od układu” wersję funkcjonału, którą nazwali SD‑SCAN. Dla materiałów kowalencyjnych — gdzie atomy dzielą elektrony w kierunkowych wiązaniach — staranne poprawki mogą przesunąć obliczone przerwy energetyczne blisko wartości doświadczalnych, zachowując jednocześnie stałe sieciowe, podstawowe wymiary kryształu, w zasadzie bez zmian. W przeciwieństwie do tego materiały silnie jonowe, gdzie elektrony są bardziej osadzone przy jednym typie atomu, pozostają trudne do poprawienia tym podejściem, co ujawnia głębsze ograniczenie leżącej u podstaw formuły.
Jak reagują elektrony i światło
Ponad samym dopasowaniem wartości przerwy, zespół bada, jak przestrojony funkcjonał zmienia szczegółową strukturę elektroniczną i rozkład elektronów między atomami. Dla ciał kowalencyjnych, takich jak diament i krzem, SD‑SCAN przesuwa nieobsadzone pasma energetyczne w górę i rozkłada ładunek bardziej realistycznie wzdłuż wiązań, wzmacniając ich charakter wspólnoelektronowy. Przy obliczaniu własności optycznych — jak materiał reaguje na światło i pola elektryczne o wysokiej częstotliwości — poprawione przerwy skutkują dokładniejszymi stałymi dielektrycznymi i lepszym dopasowaniem do mierzonych widm, szczególnie dla materiałów, gdzie oryginalny SCAN albo niedoszacowywał przerwy, albo błędnie przewidywał metaliczny charakter.
Pozwolić danym wybrać właściwe pokrętła
Ręczne strojenie parametrów dla każdego nowego materiału szybko stałoby się niepraktyczne, dlatego autorzy zwracają się ku uczeniu maszynowemu, by zautomatyzować proces. Zgromadzili większy zbiór danych obejmujący 260 półprzewodników i izolatorów i dla każdego z nich ustalili, które ustawienia parametrów działają najlepiej. Używając prostych deskryptorów — takich jak symetria krystaliczna, intensywność dzielenia lub przekazywania elektronów między atomami oraz gęstość upakowania atomów — wyszkolili modele regresyjne do przewidywania optymalnych wewnętrznych ustawień SCAN wyłącznie na podstawie tych cech fizycznych. Najlepszy model, wielokrotna regresja liniowa, potrafi oszacować przerwy z istotnie niższym błędem średnim niż standardowy SCAN, zbliżając się do ideału w pełni ręcznie dostrojonego SD‑SCAN, przy praktycznie tym samym niskim koszcie obliczeniowym.
Skrót do codziennych obliczeń
Jako jeszcze prostszą alternatywę autorzy identyfikują jeden kluczowy parametr, który szczególnie wpływa na półprzewodniki i izolatory, i proponują stałą, przestrojoną wersję nazwaną SCAN‑0.2. Ta wariant zachowuje całą fizykę SCAN, ale nieco przekształca sposób traktowania różnych środowisk wiązań. W całym ich dużym zbiorze testowym SCAN‑0.2 systematycznie poprawia przewidywania przerw energetycznych w porównaniu z oryginalnym funkcjonałem, zachowując bardzo dokładne struktury krystaliczne. W wielu układach wypada porównywalnie z podejściem opartym na uczeniu maszynowym, oferując wygodną, gotową do podmiany alternatywę do rutynowego modelowania materiałów.
Co to oznacza dla przyszłego projektowania materiałów
Mówiąc prościej, badanie pokazuje, że szeroko stosowane narzędzie teoretyczne do symulacji materiałów można uczynić zauważalnie lepszym w przewidywaniu przerw energetycznych, po prostu ucząc się, jak ustawiać jego wewnętrzne pokrętła na podstawie rodzaju badanego ciała stałego. Dla kowalencyjnych półprzewodników — głównych materiałów wykorzystywanych w elektronice — podejście dostarcza przerwy i odpowiedzi optyczne bliskie eksperymentowi przy ułamku kosztu najzaawansowanych metod. Jednocześnie pozostające trudności z materiałami silnie jonowymi podkreślają, gdzie potrzebna jest nowa fizyka, a nie tylko lepsze dopasowanie. Razem system‑zależny SD‑SCAN, oparty na danych ML‑SCAN i prosty wariant SCAN‑0.2 wskazują drogę ku przyszłości, w której receptury wymiany–korelacji nie będą już uniwersalne, lecz adaptatywnie dostosowane do każdego materiału, przyspieszając komputerowo wspomagane odkrywanie lepszych materiałów elektronicznych i fotonicznych.
Cytowanie: Dovale-Farelo, V., Tavadze, P., Marques, M.A.L. et al. System-conditioned reparameterization of the SCAN functional for accurate bandgaps: from analytical constraints to machine learning. npj Comput Mater 12, 162 (2026). https://doi.org/10.1038/s41524-026-02009-w
Słowa kluczowe: przerwy energetyczne półprzewodników, teoria funkcjonału gęstości, funkcjonał SCAN, uczenie maszynowe w nauce o materiałach, struktura elektroniczna