Clear Sky Science · fr
Reparamétrisation conditionnée au système de la fonctionnelle SCAN pour des bandgaps précis : des contraintes analytiques à l’apprentissage automatique
Pourquoi il est important d’ajuster les bandgaps
Des smartphones et des panneaux solaires aux LED et aux capteurs, de nombreuses technologies modernes reposent sur des semi‑conducteurs — des matériaux dont la capacité à conduire l’électricité est déterminée par une grandeur clé appelée bandgap. Ce bandgap est notoirement difficile à calculer avec précision sur ordinateur sans recourir à des méthodes coûteuses. L’article explore une manière plus intelligente et plus flexible d’ajuster une recette quantique‑mécanique déjà largement utilisée afin qu’elle puisse prédire les bandgaps de nombreux solides de façon plus fiable, sans payer le prix élevé des techniques les plus avancées.
Un réglage plus fin d’une théorie éprouvée
Les auteurs travaillent dans le cadre de la théorie de la fonctionnelle de la densité, une approche standard pour simuler les matériaux au niveau atomique. Dans ce cadre, l’ingrédient le plus délicat est la « fonctionnelle » échange‑corrélation, une formule qui encode la manière dont les électrons interagissent. Une version populaire, appelée SCAN, est construite à partir de contraintes physiques rigoureuses plutôt que d’un fort ajustement sur des données, ce qui la rend robuste pour de nombreuses propriétés mais seulement modérément précise pour les bandgaps. Plutôt que d’inventer une formule entièrement nouvelle, les auteurs se demandent : et si nous réajustions délicatement une poignée de boutons numériques internes dans SCAN au cas par cas pour chaque matériau, afin qu’elle reflète mieux l’environnement de liaison de chaque solide ?
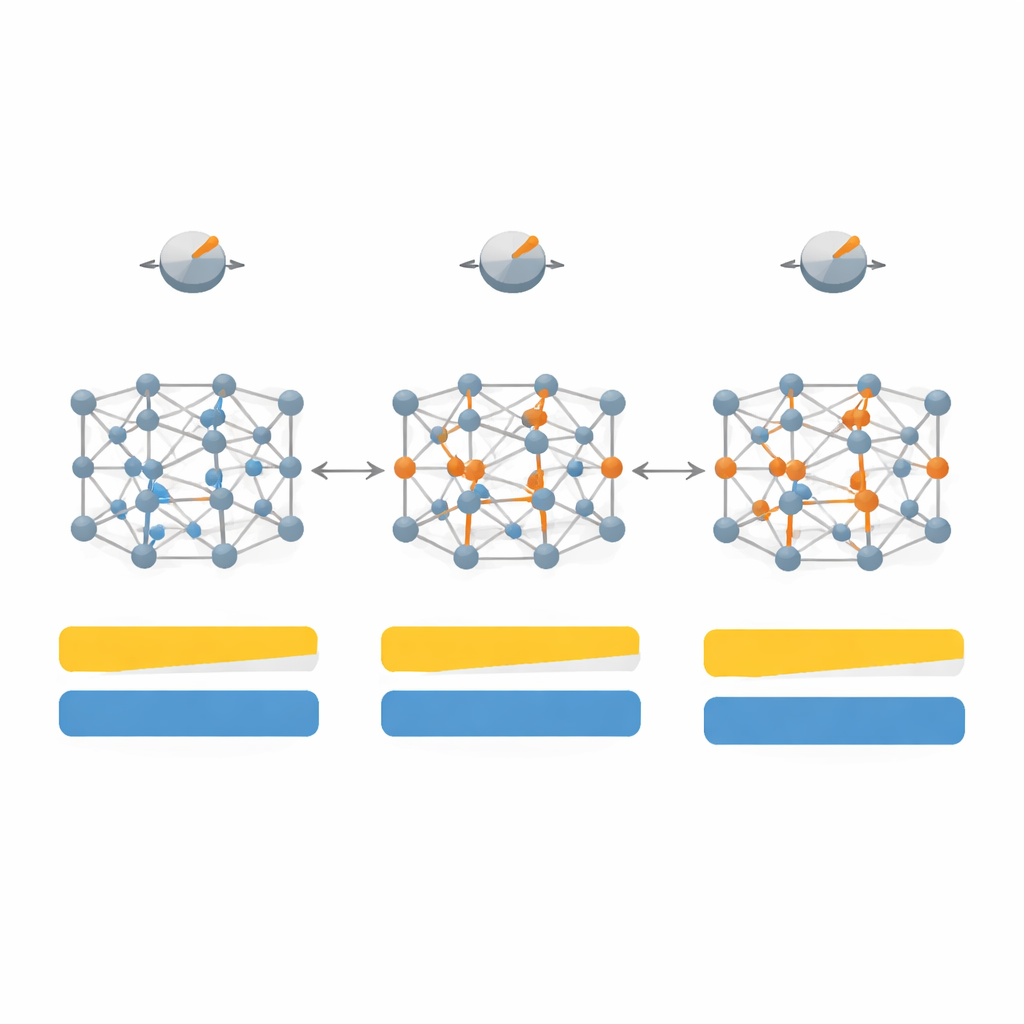
Adapter la recette à différents types de solides
Ils cartographient d’abord comment les variations de sept paramètres clés à l’intérieur de SCAN affectent à la fois le bandgap et la structure cristalline pour un ensemble de 20 matériaux représentatifs, incluant des semi‑conducteurs classiques comme le silicium et l’arséniure de gallium, des cristaux de gaz nobles inertes et des sels fortement ioniques comme le fluorure de lithium. En explorant cet espace de paramètres de haute dimension, puis en le réduisant aux quatre boutons les plus influents, ils conçoivent une version « dépendante du système » de la fonctionnelle qu’ils appellent SD‑SCAN. Pour les matériaux covalents — où les atomes partagent des électrons dans des liaisons directionnelles — des ajustements soignés peuvent rapprocher les bandgaps calculés des valeurs expérimentales tout en conservant essentiellement corrects les paramètres de maille, les dimensions de base du cristal. En revanche, les matériaux fortement ioniques, où les électrons restent plus rigidement attachés à un type d’atome, demeurent difficiles à corriger avec cette approche, révélant une limitation plus profonde de la formule sous‑jacente.
Comment réagissent les électrons et la lumière
Au‑delà de la simple concordance des valeurs de bandgap, l’équipe examine comment leur fonctionnelle réajustée modifie la structure électronique détaillée et la répartition des électrons entre atomes. Pour les solides covalents comme le diamant et le silicium, SD‑SCAN décale les bandes d’énergie vides vers des énergies plus élevées et répartit la charge de manière plus réaliste le long des liaisons, renforçant leur caractère de partage d’électrons. Lorsqu’ils calculent les propriétés optiques — la réponse du matériau à la lumière et aux champs électriques de haute fréquence — les bandgaps améliorés conduisent à des constantes diélectriques plus précises et à une meilleure concordance avec les spectres mesurés, en particulier pour les matériaux où le SCAN original sous‑estimait le gap ou prédisait à tort un comportement métallique.
Permettre aux données de choisir les bons réglages
Régler manuellement les paramètres pour chaque nouveau matériau deviendrait rapidement impraticable, aussi les auteurs se tournent‑ils vers l’apprentissage automatique pour automatiser le processus. Ils assemblent un jeu de données plus large de 260 semi‑conducteurs et isolants et, pour chacun, déterminent quels choix de paramètres fonctionnent le mieux. En utilisant des descripteurs simples — tels que la symétrie cristalline, la propension des atomes à partager ou transférer des électrons, et la compacité du réseau — ils entraînent des modèles de régression pour prédire les réglages internes optimaux de SCAN à partir de ces seules caractéristiques physiques. Leur meilleur modèle, une régression linéaire multiple, peut estimer les bandgaps avec une erreur moyenne significativement plus faible que le SCAN standard, se rapprochant de l’idéal que constitue le SD‑SCAN entièrement ajusté à la main, tout en conservant un coût informatique essentiellement aussi bas.
Un raccourci pour les calculs du quotidien
Comme alternative encore plus simple, les auteurs identifient un paramètre crucial qui affecte particulièrement les semi‑conducteurs et isolants et proposent une version réajustée fixe qu’ils appellent SCAN‑0.2. Cette variante préserve toute la physique de SCAN mais modifie légèrement la façon dont elle traite les différents environnements de liaison. Sur leur large ensemble de test, SCAN‑0.2 améliore systématiquement les prédictions de bandgap comparé à la fonctionnelle originale tout en conservant des structures cristallines très précises. Elle rivalise avec l’approche par apprentissage automatique pour de nombreux systèmes, offrant un remplacement pratique et plug‑and‑play pour la modélisation routinière des matériaux.
Ce que cela signifie pour la conception future de matériaux
En termes simples, l’étude montre qu’un outil théorique largement utilisé pour simuler les matériaux peut être sensiblement amélioré pour prédire les bandgaps simplement en apprenant à régler ses boutons internes en fonction du type de solide étudié. Pour les semi‑conducteurs covalents — les piliers de l’électronique — l’approche fournit des gaps et des réponses optiques proches de l’expérience à une fraction du coût des méthodes les plus avancées. Dans le même temps, les difficultés restantes avec les composés fortement ioniques soulignent où de nouvelles physiques, et non seulement un meilleur ajustement, sont nécessaires. Ensemble, SD‑SCAN dépendant du système, ML‑SCAN piloté par les données et la variante simple SCAN‑0.2 ouvrent la voie à un futur où les recettes échange‑corrélation ne sont plus universelles mais adaptées de manière dynamique à chaque matériau, accélérant la découverte assistée par ordinateur de meilleurs matériaux électroniques et photoniques.
Citation: Dovale-Farelo, V., Tavadze, P., Marques, M.A.L. et al. System-conditioned reparameterization of the SCAN functional for accurate bandgaps: from analytical constraints to machine learning. npj Comput Mater 12, 162 (2026). https://doi.org/10.1038/s41524-026-02009-w
Mots-clés: bandgaps des semi‑conducteurs, théorie de la fonctionnelle de la densité, fonctionnelle SCAN, apprentissage automatique en science des matériaux, structure électronique