Clear Sky Science · pt
Reparametrização condicionada ao sistema da funcional SCAN para gap de bandas precisos: de restrições analíticas ao aprendizado de máquina
Por que ajustar gaps de bandas é importante
De smartphones e painéis solares a LEDs e sensores, muitas tecnologias modernas dependem de semicondutores — materiais cuja capacidade de conduzir eletricidade é determinada por uma grandeza-chave chamada gap de banda. Esse gap é notoriamente difícil de calcular com precisão em um computador sem recorrer a métodos caros. O artigo explora uma forma mais inteligente e flexível de ajustar uma receita quântica já existente e amplamente usada para que ela preveja gaps de bandas de muitos sólidos de maneira mais confiável, sem pagar o alto custo das técnicas mais avançadas.
Um ajuste mais sutil em uma teoria confiável
Os autores trabalham dentro da teoria do funcional da densidade, uma abordagem padrão para simular materiais no nível atômico. Nesse quadro, o ingrediente mais delicado é o funcional de troca–correlação, uma fórmula que codifica como os elétrons interagem. Uma versão popular chamada SCAN é construída a partir de restrições físicas rigorosas em vez de um grande ajuste a dados, o que a torna robusta para muitas propriedades, mas apenas moderadamente precisa para gaps de bandas. Em vez de inventar uma fórmula totalmente nova, os autores perguntam: e se retocarmos suavemente um punhado de controles numéricos internos no SCAN material a material, de modo que ele reflita melhor o ambiente de ligação de cada sólido?
Ajustando a receita para diferentes tipos de sólidos
Eles primeiramente mapeiam como mudanças em sete parâmetros-chave dentro do SCAN afetam tanto o gap de banda quanto a estrutura cristalina para um conjunto de 20 materiais representativos, incluindo semicondutores clássicos como silício e arseneto de gálio, cristais inertes de gases nobres e sais fortemente iônicos como fluoreto de lítio. Ao explorar esse espaço de parâmetros de alta dimensão e então reduzi-lo aos quatro botões mais influentes, projetam uma versão "dependente do sistema" do funcional que chamam SD‑SCAN. Para materiais covalentes — onde átomos compartilham elétrons em ligações direcionais — ajustes cuidadosos podem aproximar os gaps calculados dos valores experimentais mantendo, ao mesmo tempo, as constantes de rede, as dimensões básicas do cristal, essencialmente corretas. Em contraste, materiais altamente iônicos, onde os elétrons ficam mais rigidamente presos a um tipo de átomo, continuam difíceis de corrigir com essa abordagem, revelando uma limitação mais profunda da fórmula subjacente.
Como os elétrons e a luz respondem
Além de apenas igualar números de gaps, a equipe examina como seu funcional retocado altera a estrutura eletrônica detalhada e como os elétrons são distribuídos entre os átomos. Para sólidos covalentes como diamante e silício, o SD‑SCAN desloca as bandas vazias de energia para cima e distribui a carga de forma mais realista ao longo das ligações, reforçando seu caráter de elétrons compartilhados. Quando calculam propriedades ópticas — como o material responde à luz e a campos elétricos de alta frequência — os gaps melhorados conduzem a constantes dielétricas mais precisas e melhor concordância com espectros medidos, especialmente para materiais onde o SCAN original subestimava o gap ou até previu erroneamente comportamento metálico.
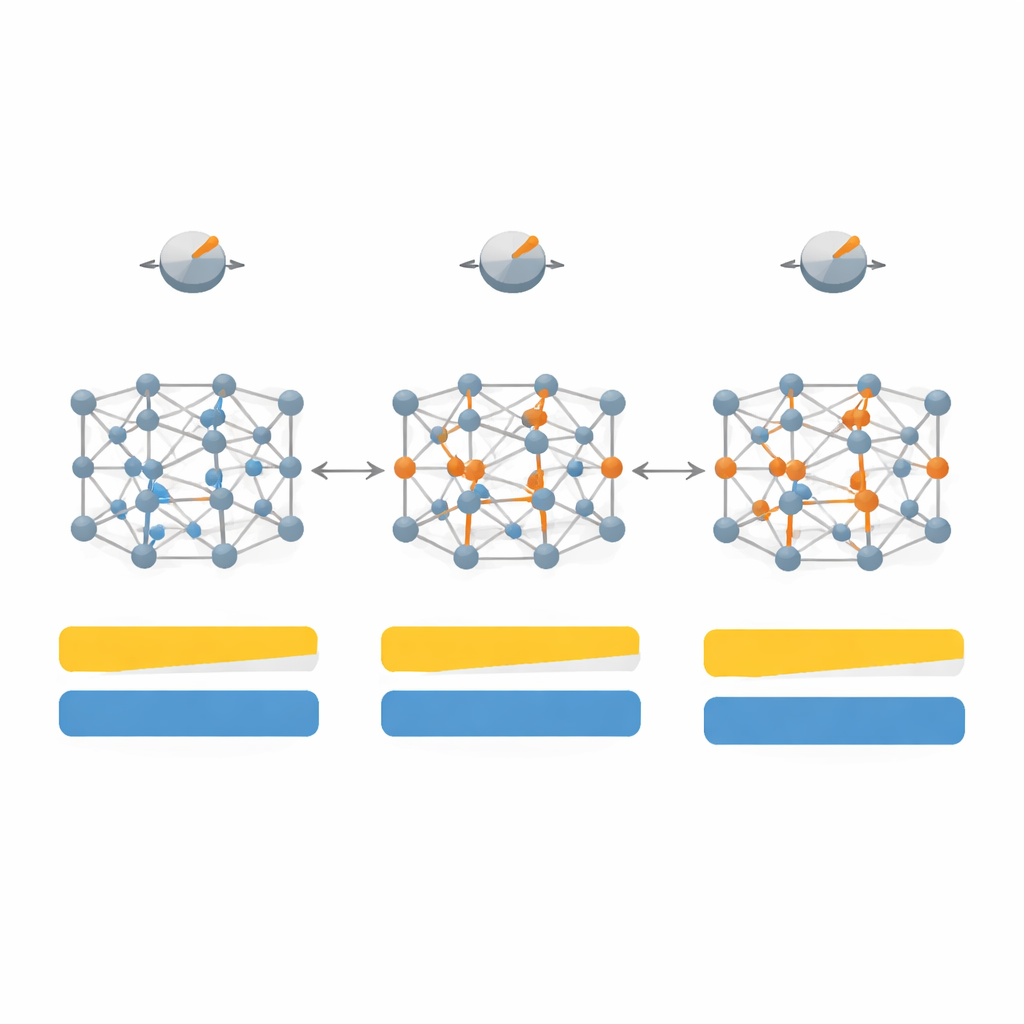
Deixando os dados escolherem os botões certos
Ajustar manualmente os parâmetros para cada novo material rapidamente se tornaria impraticável, então os autores recorrem ao aprendizado de máquina para automatizar o processo. Eles reúnem um conjunto de dados maior, com 260 semicondutores e isolantes e, para cada um, determinam quais escolhas de parâmetros funcionam melhor. Usando descritores simples — como simetria cristalina, o quão fortemente os átomos compartilham ou transferem elétrons e quão compactos os átomos estão — treinam modelos de regressão para prever as configurações internas ótimas do SCAN a partir apenas dessas características físicas. Seu melhor modelo, uma regressão linear múltipla, consegue estimar gaps de bandas com erro médio significativamente menor que o SCAN padrão, aproximando‑se do ideal SD‑SCAN totalmente ajustado à mão, porém com essencialmente o mesmo baixo custo computacional.
Um atalho para cálculos do dia a dia
Como alternativa ainda mais simples, os autores identificam um parâmetro crucial que afeta particularmente semicondutores e isolantes e propõem uma versão re‑ajustada fixa que chamam SCAN‑0.2. Essa variante mantém toda a física do SCAN intacta, mas molda ligeiramente como ele trata diferentes ambientes de ligação. Em seu amplo conjunto de testes, o SCAN‑0.2 melhora sistematicamente as previsões de gap em comparação com o funcional original, mantendo estruturas cristalinas muito precisas. Ele tem desempenho comparável à abordagem por aprendizado de máquina para muitos sistemas, oferecendo um substituto conveniente plug‑and‑play para modelagem rotineira de materiais.
O que isso significa para o futuro do projeto de materiais
Em termos simples, o estudo mostra que uma ferramenta teórica amplamente usada para simular materiais pode ser significativamente melhorada na previsão de gaps de bandas simplesmente aprendendo a ajustar seus botões internos com base no tipo de sólido estudado. Para semicondutores covalentes — as bases da eletrônica — a abordagem fornece gaps e respostas ópticas próximos ao experimento a uma fração do custo dos métodos mais avançados. Ao mesmo tempo, as dificuldades remanescentes com compostos altamente iônicos sublinham onde física nova, e não apenas melhor ajuste, é necessária. Em conjunto, o SD‑SCAN dependente do sistema, o ML‑SCAN guiado por dados e a simples variante SCAN‑0.2 apontam para um futuro em que receitas de troca–correlação não são mais de tamanho único, mas adaptadas a cada material, acelerando a descoberta por computador de materiais eletrônicos e fotônicos melhores.
Citação: Dovale-Farelo, V., Tavadze, P., Marques, M.A.L. et al. System-conditioned reparameterization of the SCAN functional for accurate bandgaps: from analytical constraints to machine learning. npj Comput Mater 12, 162 (2026). https://doi.org/10.1038/s41524-026-02009-w
Palavras-chave: gaps de bandas em semicondutores, teoria do funcional da densidade, funcional SCAN, aprendizado de máquina em ciência dos materiais, estrutura eletrônica