Clear Sky Science · it
Riparametrizzazione condizionata dal sistema della funzione SCAN per gap di banda accurati: dai vincoli analitici al machine learning
Perché è importante tarare i gap di banda
Dagli smartphone e i pannelli solari ai LED e ai sensori, molte tecnologie moderne dipendono dai semiconduttori — materiali la cui capacità di condurre elettricità è determinata da una grandezza chiave chiamata gap di banda. Questo gap è notoriamente difficile da calcolare con precisione su un computer senza ricorrere a metodi costosi. L’articolo esplora un modo più intelligente e flessibile per ritoccare una ricetta quantomeccanica esistente e ampiamente usata, così che possa prevedere i gap di banda di molti solidi in modo più affidabile, senza pagare il costo elevato delle tecniche più avanzate.
Una manopola più intelligente su una teoria affidabile
Gli autori lavorano nell’ambito della teoria del funzionale della densità, un approccio standard per simulare i materiali a livello atomico. In questo quadro, l’ingrediente più delicato è il funzionale di scambio‑correlazione, una formula che codifica come interagiscono gli elettroni. Una versione popolare chiamata SCAN è costruita su vincoli fisici rigorosi piuttosto che su un ampio fitting ai dati, il che la rende robusta per molte proprietà ma solo moderatamente accurata per i gap di banda. Invece di inventare una formula completamente nuova, gli autori si chiedono: e se ritoccassimo delicatamente una manciata di manopole numeriche interne in SCAN per ciascun materiale, in modo che rifletta meglio l’ambiente di legame di ogni solido?
Regolare la ricetta per diversi tipi di solidi
Per prima cosa mappano come le variazioni in sette parametri chiave all’interno di SCAN influenzano sia il gap di banda sia la struttura cristallina per un insieme di 20 materiali rappresentativi, includendo semiconduttori classici come silicio e arsenide di gallio, cristalli di gas nobili inerti e sali fortemente ionici come il fluoruro di litio. Esplorando questo spazio parametrico ad alta dimensione, e poi restringendolo alle quattro manopole più influenti, progettano una versione “dipendente dal sistema” del funzionale che chiamano SD‑SCAN. Per materiali covalenti — dove gli atomi condividono elettroni in legami direzionali — regolazioni attente possono portare i gap calcolati vicino ai valori sperimentali pur mantenendo essenzialmente corretti i costanti reticolari, le dimensioni di base del cristallo. Al contrario, i materiali altamente ionici, dove gli elettroni sono più rigidamente localizzati su un tipo di atomo, restano difficili da correggere con questo approccio, rivelando una limitazione più profonda della formula di base.
Come rispondono elettroni e luce
Oltre a far corrispondere i numeri dei gap, il gruppo esamina come il funzionale ritoccato modifichi la struttura elettronica dettagliata e la distribuzione di carica tra gli atomi. Per solidi covalenti come il diamante e il silicio, SD‑SCAN sposta verso l’alto le bande vuote di energia e distribuisce la carica in modo più realistico lungo i legami, rafforzandone il carattere di condivisione elettronica. Quando calcolano le proprietà ottiche — come il materiale risponde alla luce e ai campi elettrici ad alta frequenza — i gap migliorati conducono a costanti dielettriche più accurate e a un miglior accordo con spettri misurati, specialmente per materiali dove l’SCAN originale sottostimava il gap o addirittura prediceva erroneamente un comportamento metallico.
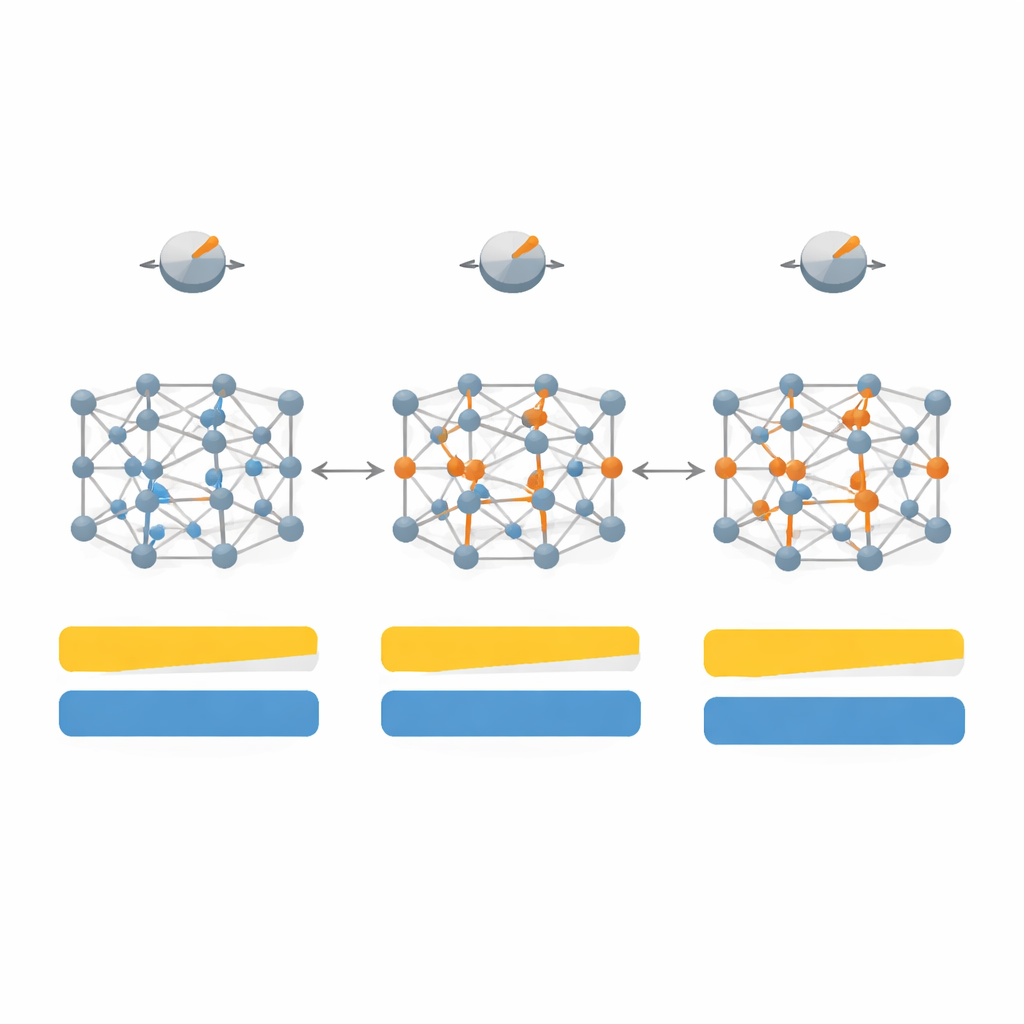
Lasciare che i dati scelgano le manopole giuste
Tarare manualmente i parametri per ogni nuovo materiale diventerebbe rapidamente impraticabile, quindi gli autori ricorrono al machine learning per automatizzare il processo. Assemblano un dataset più ampio di 260 semiconduttori e isolanti e, per ciascuno, determinano quali scelte di parametri funzionano meglio. Usando descrittori semplici — come la simmetria cristallina, quanto gli atomi condividono o trasferiscono elettroni e quanto sono densamente impaccati — addestrano modelli di regressione per prevedere le impostazioni interne ottimali di SCAN a partire solo da queste caratteristiche fisiche. Il loro miglior modello, una regressione lineare multipla, può stimare i gap con un errore medio significativamente inferiore rispetto allo SCAN standard, avvicinandosi all’ideale SD‑SCAN completamente tarato a mano, ma a un costo computazionale sostanzialmente pari a quello basso di SCAN.
Una scorciatoia per i calcoli di tutti i giorni
Come alternativa ancora più semplice, gli autori identificano un parametro cruciale che influisce in modo particolare su semiconduttori e isolanti e propongono una versione fissa ritoccata che chiamano SCAN‑0.2. Questa variante mantiene intatta tutta la fisica di SCAN ma rimodella lievemente il trattamento dei diversi ambienti di legame. Sul loro ampio set di test, SCAN‑0.2 migliora sistematicamente le previsioni dei gap rispetto al funzionale originale mantenendo strutture cristalline molto accurate. Per molti sistemi si comporta alla pari con l’approccio basato sul machine learning, offrendo un comodo sostituto plug‑in per la modellizzazione routinaria dei materiali.
Cosa significa per il futuro della progettazione dei materiali
In termini semplici, lo studio mostra che uno strumento teorico ampiamente usato per simulare materiali può essere reso significativamente migliore nella previsione dei gap di banda semplicemente imparando a impostare le sue manopole interne in base al tipo di solido studiato. Per i semiconduttori covalenti — i cavalli da lavoro dell’elettronica — l’approccio fornisce gap e risposte ottiche vicini all’esperimento a una frazione del costo dei metodi più avanzati. Allo stesso tempo, le difficoltà residue con i composti altamente ionici indicano dove è necessaria nuova fisica, non solo un migliore fitting. Insieme, SD‑SCAN dipendente dal sistema, ML‑SCAN guidato dai dati e la semplice variante SCAN‑0.2 indicano un futuro in cui le ricette di scambio‑correlazione non sono più taglia unica, ma sono adattate a ciascun materiale, accelerando la scoperta guidata al computer di materiali elettronici e fotonici migliori.
Citazione: Dovale-Farelo, V., Tavadze, P., Marques, M.A.L. et al. System-conditioned reparameterization of the SCAN functional for accurate bandgaps: from analytical constraints to machine learning. npj Comput Mater 12, 162 (2026). https://doi.org/10.1038/s41524-026-02009-w
Parole chiave: gap di banda nei semiconduttori, teoria del funzionale della densità, funzionale SCAN, machine learning nella scienza dei materiali, struttura elettronica