Clear Sky Science · es
Reparametrización condicionada al sistema del funcional SCAN para brechas de banda precisas: desde restricciones analíticas hasta aprendizaje automático
Por qué importa ajustar las brechas de banda
Desde teléfonos inteligentes y paneles solares hasta LEDs y sensores, muchas tecnologías actuales dependen de semiconductores —materiales cuya capacidad para conducir electricidad está determinada por una magnitud clave llamada brecha de banda. Esta brecha es notoriamente difícil de calcular con precisión en un ordenador sin recurrir a métodos costosos. El artículo explora una forma más inteligente y flexible de ajustar una receta cuántico‑mecánica existente y muy utilizada para que pueda predecir las brechas de banda de numerosos sólidos con mayor fiabilidad, sin asumir el elevado coste de las técnicas más avanzadas.
Un mando más listo sobre una teoría de confianza
Los autores trabajan dentro de la teoría del funcional de la densidad, un enfoque estándar para simular materiales a escala atómica. En este marco, el ingrediente más delicado es el «funcional» de intercambio‑correlación, una fórmula que codifica cómo interactúan los electrones. Una versión popular llamada SCAN se construye a partir de restricciones físicas rigurosas en lugar de un ajuste intensivo a datos, lo que la hace robusta para muchas propiedades pero solo moderadamente precisa para las brechas de banda. En lugar de inventar una fórmula completamente nueva, los autores plantean: ¿y si retocamos suavemente un puñado de perillas numéricas internas en SCAN caso por caso, de modo que refleje mejor el entorno de enlace de cada sólido?
Ajustando la receta para distintos tipos de sólidos
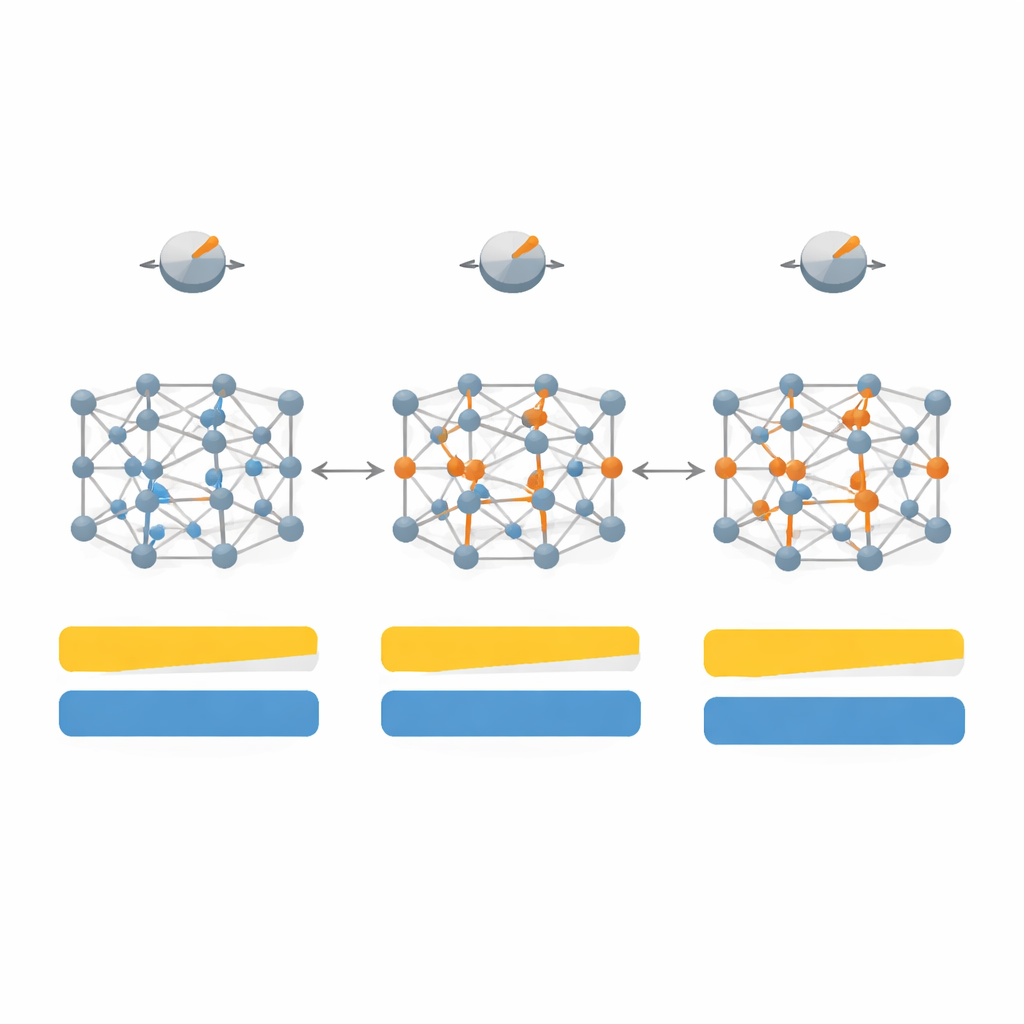
Primero cartografían cómo los cambios en siete parámetros clave dentro de SCAN afectan tanto a la brecha de banda como a la estructura cristalina para un conjunto de 20 materiales representativos, incluidos semiconductores clásicos como silicio y arseniuro de galio, cristales de gases nobles inertes y sales fuertemente iónicas como el fluoruro de litio. Al explorar este espacio de parámetros de alta dimensión y luego reducirlo a las cuatro perillas más influyentes, diseñan una versión «dependiente del sistema» del funcional que denominan SD‑SCAN. Para materiales covalentes —donde los átomos comparten electrones en enlaces direccionales— los ajustes cuidadosos pueden acercar las brechas de banda calculadas a los valores experimentales sin dejar de mantener esenciales correctas las constantes de red, las dimensiones básicas del cristal. En contraste, los materiales altamente iónicos, donde los electrones permanecen más rígidamente sobre un tipo de átomo, siguen siendo difíciles de corregir con este enfoque, lo que revela una limitación más profunda de la fórmula subyacente.
Cómo responden los electrones y la luz
Más allá de ajustar solo los números de la brecha, el equipo examina cómo su funcional retocado cambia la estructura electrónica detallada y cómo se distribuyen los electrones entre los átomos. Para sólidos covalentes como el diamante y el silicio, SD‑SCAN desplaza las bandas vacías hacia arriba y distribuye la carga de forma más realista a lo largo de los enlaces, reforzando su carácter de electrones compartidos. Cuando calculan propiedades ópticas —cómo responde el material a la luz y a campos eléctricos de alta frecuencia— las brechas mejoradas conducen a constantes dieléctricas más precisas y a un mejor acuerdo con espectros medidos, especialmente en materiales donde el SCAN original subestimaba la brecha o incluso predecía erróneamente un comportamiento metálico.
Dejar que los datos elijan las perillas adecuadas
Ajustar manualmente parámetros para cada nuevo material resultaría rápidamente impracticable, por lo que los autores recurren al aprendizaje automático para automatizar el proceso. Reúnen un conjunto de datos mayor de 260 semiconductores e aislantes y, para cada uno, determinan qué elecciones de parámetros funcionan mejor. Usando descriptores sencillos —como la simetría cristalina, la tendencia de los átomos a compartir o transferir electrones y la densidad de empaquetamiento atómico— entrenan modelos de regresión para predecir las configuraciones internas óptimas de SCAN a partir de estas características físicas. Su mejor modelo, una regresión lineal múltiple, puede estimar brechas de banda con un error medio significativamente menor que el SCAN estándar, acercándose al ideal SD‑SCAN totalmente ajustado a mano, pero al mismo coste computacional reducido.
Un atajo para cálculos cotidianos
Como alternativa aún más simple, los autores identifican un parámetro crucial que afecta especialmente a semiconductores e aislantes y proponen una versión re‑ajustada fija que llaman SCAN‑0.2. Esta variante conserva toda la física de SCAN pero modifica ligeramente cómo trata distintos entornos de enlace. En su amplio conjunto de pruebas, SCAN‑0.2 mejora sistemáticamente las predicciones de brechas de banda respecto al funcional original sin sacrificar la precisión de las estructuras cristalinas. Rinde a la par con el enfoque de aprendizaje automático para muchos sistemas, ofreciendo un reemplazo cómodo y listo para usar en modelización rutinaria de materiales.
Qué significa esto para el diseño futuro de materiales
En términos sencillos, el estudio muestra que una herramienta teórica ampliamente usada para simular materiales puede hacerse notablemente mejor en la predicción de brechas de banda simplemente aprendiendo a ajustar sus mandos internos según el tipo de sólido estudiado. Para semiconductores covalentes —los caballos de batalla de la electrónica— el enfoque ofrece brechas y respuestas ópticas cercanas al experimento a una fracción del coste de los métodos más avanzados. Al mismo tiempo, las dificultades persistentes con compuestos muy iónicos subrayan dónde se necesita nueva física, no solo un mejor ajuste. En conjunto, SD‑SCAN dependiente del sistema, ML‑SCAN basado en datos y la simple variante SCAN‑0.2 apuntan hacia un futuro en el que las recetas de intercambio‑correlación dejan de ser de talla única y se adaptan a cada material, acelerando el descubrimiento asistido por ordenador de mejores materiales electrónicos y fotónicos.
Cita: Dovale-Farelo, V., Tavadze, P., Marques, M.A.L. et al. System-conditioned reparameterization of the SCAN functional for accurate bandgaps: from analytical constraints to machine learning. npj Comput Mater 12, 162 (2026). https://doi.org/10.1038/s41524-026-02009-w
Palabras clave: brechas de banda en semiconductores, teoría del funcional de la densidad, funcional SCAN, aprendizaje automático en ciencia de materiales, estructura electrónica