Clear Sky Science · sv
Spatial single cell transcriptomic analysis informs tumor developmental hierarchy of DICER1 syndrome related sarcoma
Varför dessa ovanliga barndomscancerformer är viktiga
DICER1‑syndromet är en sällsynt ärftlig åkomma som ökar risken för cancer, särskilt hos barn och unga vuxna. Många av dessa tumörer är sarkom, cancer som växer från kroppens stödjevävnad. Trots att de kan dyka upp i olika organ ser de slående lika ut i mikroskopet. I den här studien ställs en grundläggande men viktig fråga: var börjar dessa cancerformer och hur förändras de när de växer?
Närmare titt på ett cancerpredispositionssyndrom
Personer med DICER1‑syndrom föds med en skadlig förändring i en kopia av DICER1‑genen, som hjälper till att bearbeta små RNA‑molekyler som finjusterar många andra gener. Tumörer utvecklas vanligtvis först efter att den andra DICER1‑kopian fått ett specifikt fel i ett område som kallas RNase IIIb‑domän. Detta rubbar balansen av små RNA och genaktivitet. Trots att tumörerna kan uppträda i lunga, njure, hjärna och fortplantningsorgan delar många av dem liknande strukturer och beteenden, vilket antyder att de kan ha sitt ursprung i en gemensam celltyp.
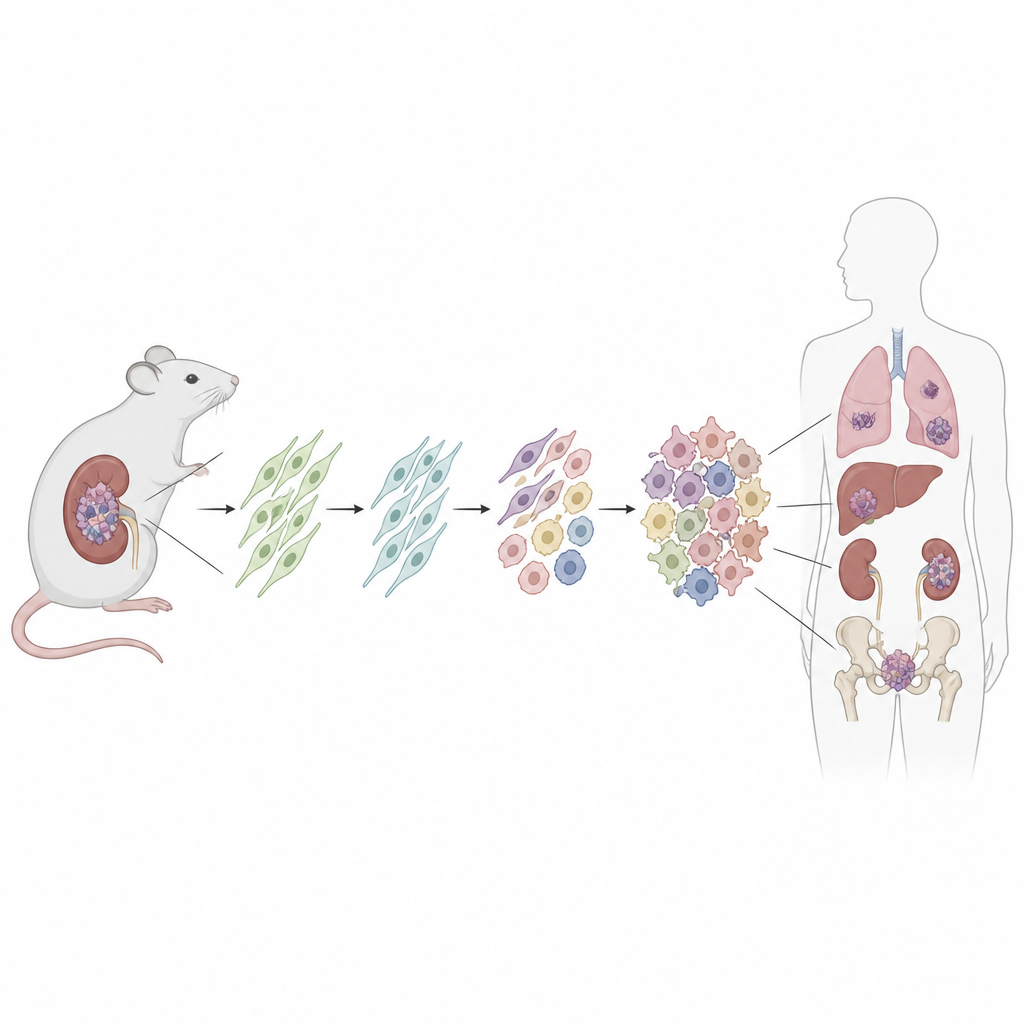
Att bygga en spårbar cancer i mus
För att leta efter ursprungscellen konstruerade forskarna en mus där en patientlik Dicer1‑mutation kan aktiveras i en definierad uppsättning i vila befintliga stödjeceller kallade mesenkymala stromaceller. De använde en läkemedelstriggad aktivering efter födseln och slog också på en fluorescerande tagg för att permanent märka alla avkommaceller. Med tiden utvecklade de flesta av dessa möss njurtumörer som starkt påminde om mänskliga DICER1‑relaterade sarkom, från tidiga låggradiga lesioner till stora, aggressiva massor som överskred njurens gränser. Omgivande njurstrukturer, som beklädnaden av cystor, bar inte den fluorescerande taggen, vilket visade att de inte själva var cancerösa.
Att hitta en sårbar fibroblastförfader
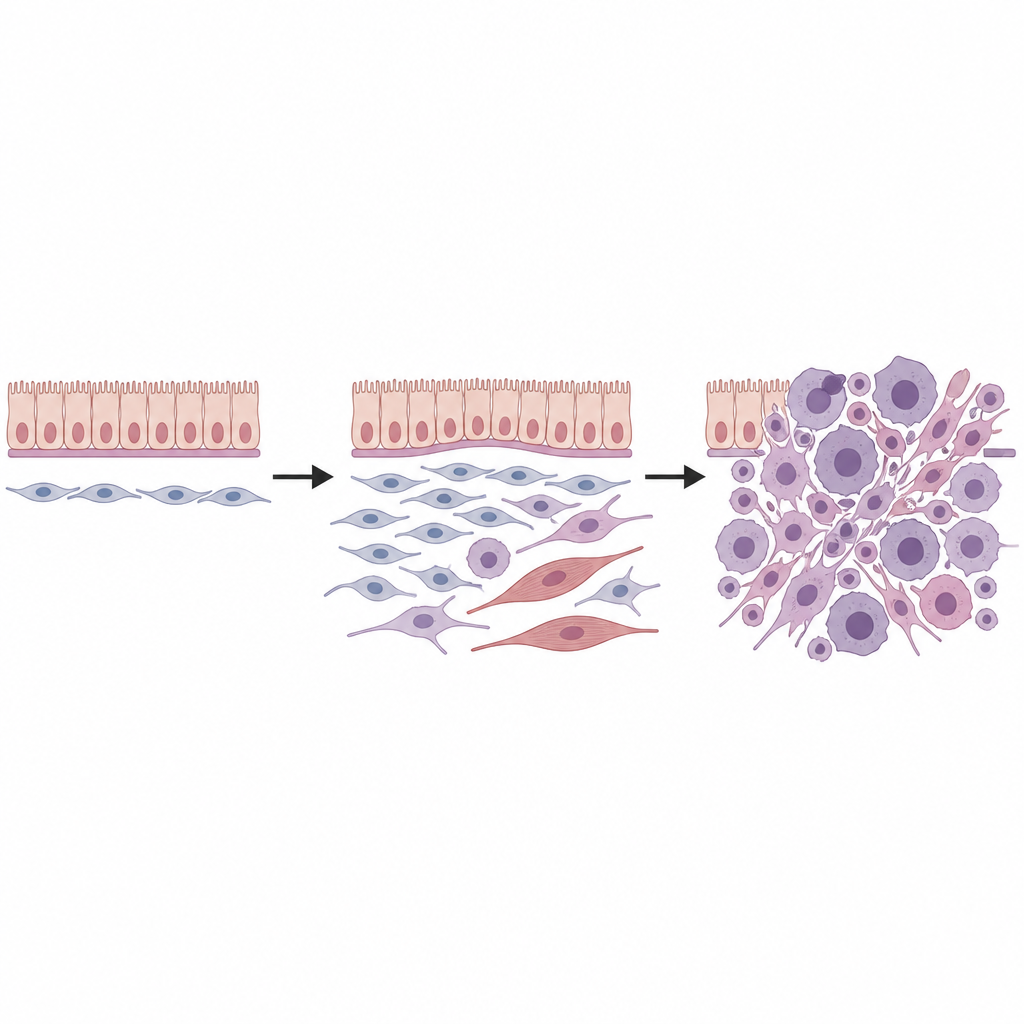
Därefter kombinerade teamet single cell‑RNA‑sekvensering, som avläser genaktivitet cell för cell, med spatial transkriptomik, som kartlägger dessa celler tillbaka i vävnadssektioner. I friska njurar fann de flera typer mesenkymala celler, inklusive en särskild grupp fibroblaster belägna runt blodkärl, under ytskikt och under övergångsepitel i samlingsgångarna. Dessa så kallade universella fibroblaster, markerade av en specifik genblandning, verkade sitta i roten av ett litet stamträd för stödjeceller. När Dicer1‑mutationen aktiverades omprogrammerades dessa fibroblaster: de förlorade markörer för mogna linjer och fick kännetecken av ett mer primitivt, flexibelt tillstånd som var redo för malign förändring.
Att följa tumörutveckling cell för cell
Genom att följa tusentals individuella celler med hjälp av beräkningsmässig ”pseudotime”‑analys rekonstruerade forskarna flera utvecklingsvägar som utgick från dessa universella fibroblaster. En väg ledde mot celler som började likna omoget muskelvävnad och sedan fullt utvecklade skelettmuskel‑lika celler, vilket speglar den rhabdomyoblasta utseendet som ses i många DICER1‑sarkom. En annan bana gav fibroblastlika tumörceller som samlade kromosomala vinster, särskilt involverande muschromosomerna 1 och 6, för att sedan utvecklas till starkt prolifererande sarkomceller. Ytterligare genetiska träffar, inklusive mutationer i klassiska cancergener som p53 och Kras, drev tillväxt och aggressivt beteende vidare, delvis genom att dämpa cellernas dödsskydd och öka MAPK‑signalering.
Att knyta ihop mus‑ och människotumörer
För att undersöka om fynden är relevanta för människor tillämpade teamet spatial transkriptomik på sexton mänskliga DICER1‑relaterade mesenkymala tumörer från olika organ och kliniska undertyper. De fann återigen ett kärnset av celltillstånd: en progenitorpopulation som delade den universella fibroblastens gensignatur, prolifererande tumörceller, mellanliggande och fullt muskel‑lika celler samt en bakgrund av mer ordinära fibroblaster. Den rumsliga ordningen av dessa celler, inklusive ett kompakt ”cambiumlager” av progenitorer under epitelbeklädnader, återspeglade tydligt muskmodellen. Överensstämmelsen med kända cellprogram i barndomsrhabdomyosarkom antyder att åtminstone några av dessa muskel‑lika cancerformer kan härstamma från fibroblaster snarare än från redan förpliktade muskelprekursorer.
Vad detta betyder för patienter och framtida behandlingar
Detta arbete ger starka bevis för att DICER1‑syndromsrelaterade sarkom uppstår från en gemensam fibroblastförfader som normalt stödjer vävnader nära epitelytor och blodkärl. När DICER1‑funktionen störs i dessa celler blir de ovanligt plastiska: vissa rör sig mot en muskel‑lik identitet medan andra utvecklas till snabbt växande sarkomceller när de samlar på sig ytterligare genetiska skador. Genom att bygga en trogen och spårbar musemodell och jämföra den med mänskliga tumörer lägger studien grunden för att dissekera tidiga sjukdomssteg och testa terapier som riktar in sig på nyckelvägar för tillväxt, såsom MAPK‑signalering, innan dessa sällsynta men allvarliga cancerformer fullt ut tar fäste.
Citering: Kommoss, F.K.F., Zhang, J.Y.H., Lynch, B.J. et al. Spatial single cell transcriptomic analysis informs tumor developmental hierarchy of DICER1 syndrome related sarcoma. Nat Commun 17, 4608 (2026). https://doi.org/10.1038/s41467-026-70971-6
Nyckelord: DICER1‑syndrom, sarkom, fibroblaster, single cell‑transkriptomik, musmodell för cancer