Clear Sky Science · it
Analisi trascrittomica spaziale a singola cellula illumina la gerarchia di sviluppo dei tumori nella sindrome DICER1 correlata al sarcoma
Perché questi rari tumori infantili sono importanti
La sindrome DICER1 è una rara condizione ereditaria che aumenta il rischio di sviluppare tumori, soprattutto nei bambini e nei giovani adulti. Molti di questi tumori sono sarcomi, neoplasie che derivano dai tessuti di supporto dell’organismo. Sebbene possano comparire in organi diversi, al microscopio si somigliano in modo sorprendente. Questo studio pone una domanda semplice ma cruciale: da dove iniziano questi tumori e come cambiano durante la loro crescita?
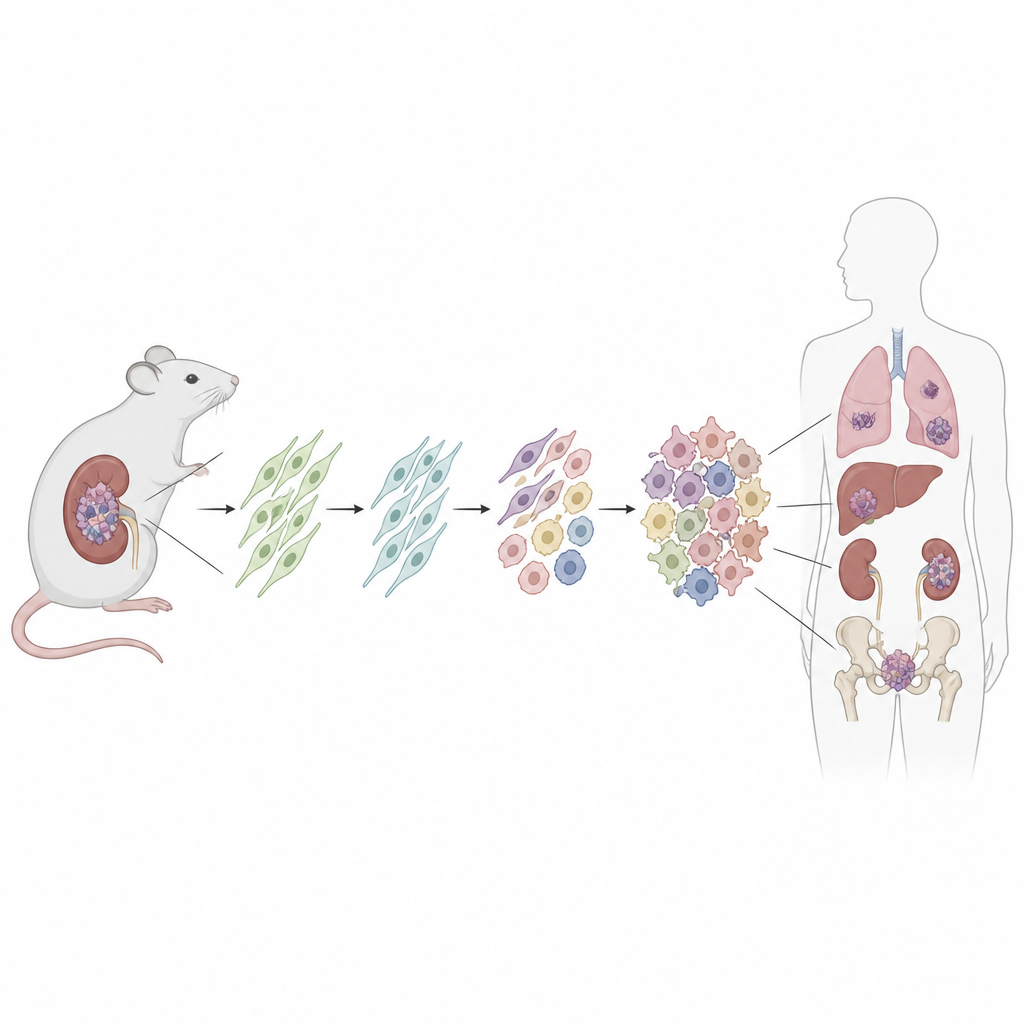
Uno sguardo più ravvicinato a una sindrome di predisposizione al cancro
Le persone con sindrome DICER1 nascono con una variazione dannosa in una copia del gene DICER1, che contribuisce a elaborare piccole molecole di RNA che modulano molti altri geni. I tumori di solito si sviluppano dopo che la seconda copia di DICER1 acquisisce un difetto specifico in una regione chiamata dominio RNase IIIb. Ciò altera l’equilibrio normale degli small RNA e l’attività genica. Nonostante compaiano nel polmone, nel rene, nel cervello e negli organi riproduttivi, molti di questi tumori condividono strutture e comportamenti simili, suggerendo che possano originare da un tipo cellulare comune.
Costruire un cancro tracciabile in un topo
Per cercare la cellula d’origine, i ricercatori hanno ingegnerizzato un topo in cui una mutazione di Dicer1 simile a quella dei pazienti può essere attivata in un insieme definito di cellule di supporto quiescenti chiamate cellule stromali mesenchimali. Hanno usato un farmaco per scatenare la mutazione dopo la nascita e hanno anche attivato un marcatore fluorescente per etichettare in modo permanente tutte le cellule discendenti. Col tempo, nella maggior parte di questi topi si sono sviluppati tumori renali che rispecchiavano da vicino i sarcomi umani correlati a DICER1, da lesioni precoci a basso grado fino a masse ampie e aggressive che invadevano il rene. Le strutture renali circostanti, come l’epitelio delle cisti, non portavano il marcatore fluorescente, dimostrando che non erano esse stesse cancerose.
Trovare un antenato fibroblastico vulnerabile
Successivamente il gruppo ha combinato il sequenziamento dell’RNA a singola cellula, che misura l’attività genica cellula per cellula, con la trascrittomica spaziale, che rimappa quelle cellule nelle sezioni tissutali. Nei reni sani hanno identificato diversi tipi di cellule mesenchimali, inclusi un gruppo speciale di fibroblasti localizzati intorno ai vasi sanguigni, sotto i rivestimenti superficiali e al di sotto dell’epitelio di transizione dei dotti collettori. Questi cosiddetti fibroblasti universali, contrassegnati da un mix specifico di geni, sembravano trovarsi alla radice di un piccolo albero genealogico di cellule di supporto. Quando la mutazione di Dicer1 è stata attivata, questi fibroblasti sono stati riprogrammati: hanno perso marcatori delle linee mature e hanno acquisito caratteristiche di uno stato più primitivo e flessibile, predisposto al cambiamento maligno.
Osservare l’evoluzione dei tumori cellula per cellula
Seguendo migliaia di singole cellule tramite analisi computazionale di “pseudotempo”, i ricercatori hanno ricostruito diversi percorsi di sviluppo a partire da questi fibroblasti universali. Una via ha portato a cellule che cominciavano ad assomigliare a cellule muscolari immature e poi a cellule completamente simili al muscolo scheletrico, rispecchiando l’aspetto rabdomioblastico osservato in molti sarcomi DICER1. Un’altra via ha prodotto cellule tumorali di tipo fibroblastico che accumulavano guadagni cromosomici, in particolare coinvolgendo i cromosomi murini 1 e 6, e poi sono progredite in cellule sarcomatose altamente proliferative. Colpi genetici aggiuntivi, incluse mutazioni in geni classici del cancro come p53 e Kras, hanno ulteriormente alimentato la crescita e il comportamento aggressivo, in parte attenuando i meccanismi di morte cellulare e potenziando le vie di segnalazione MAPK.
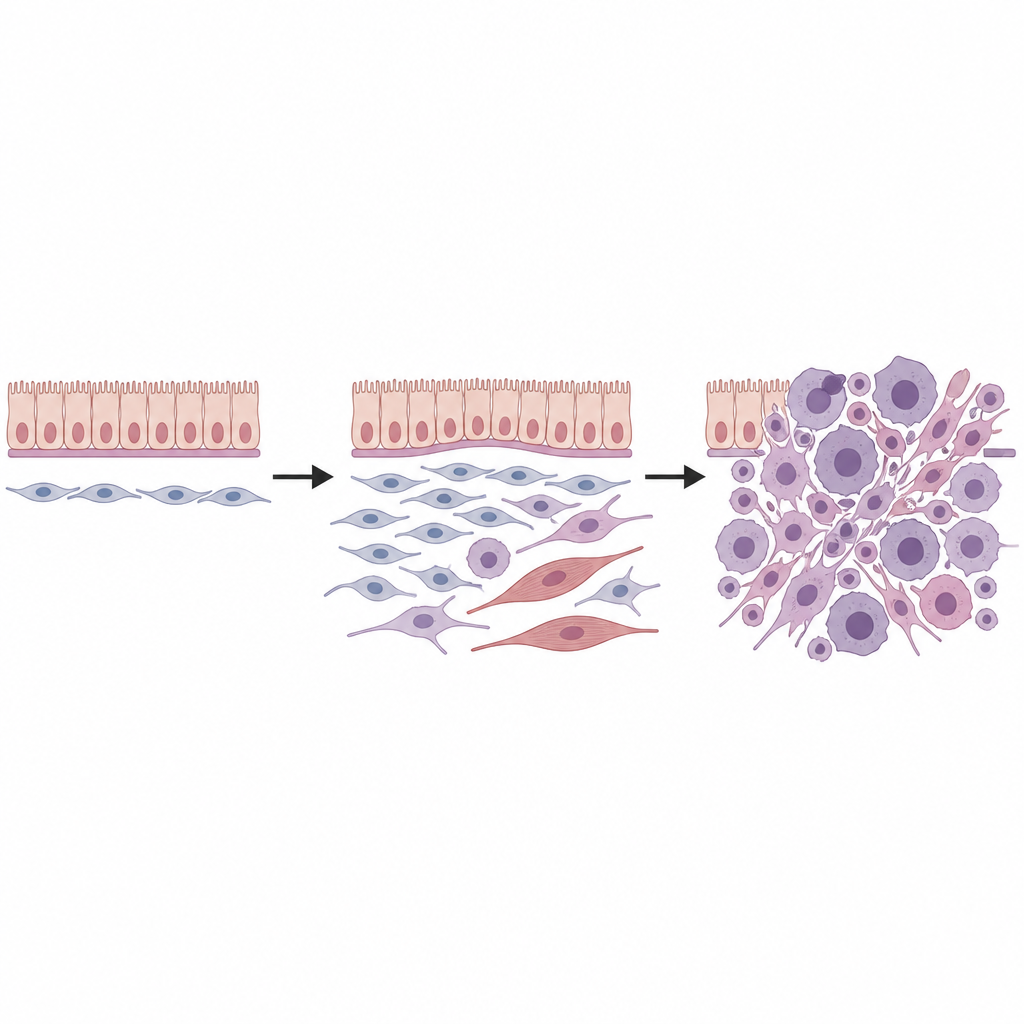
Mettere in relazione i tumori del topo e dell’uomo
Per verificare se queste osservazioni fossero rilevanti per l’uomo, il gruppo ha applicato la trascrittomica spaziale a sedici tumori mesenchimali umani correlati a DICER1 provenienti da vari organi e sottotipi clinici. Hanno nuovamente scoperto un nucleo di stati cellulari: una popolazione progenitrice che condivideva la firma genica dei fibroblasti universali, cellule tumorali proliferanti, cellule intermedie e completamente simili al muscolo, e uno sfondo di fibroblasti più ordinari. L’organizzazione spaziale di queste cellule, inclusa uno spesso “strato cambiale” di progenitori sotto i rivestimenti epiteliali, rispecchiava da vicino i tumori murini. La sovrapposizione con programmi cellulari noti nel rabdomiosarcoma infantile suggerisce che almeno alcune di queste neoplasie muscolari potrebbero derivare da fibroblasti piuttosto che da precursori muscolari già impegnati.
Cosa significa per i pazienti e per le terapie future
Questo lavoro fornisce solide prove che i sarcomi correlati alla sindrome DICER1 originano da un antenato fibroblastico comune che normalmente supporta i tessuti vicino alle superfici epiteliali e ai vasi sanguigni. Quando la funzione di DICER1 viene alterata in queste cellule, esse diventano insolitamente plastiche: alcune evolvono verso un’identità simile al muscolo, mentre altre progrediscono in cellule sarcomatose a rapida crescita accumulando ulteriori danni genetici. Costruendo un modello murino fedele e tracciabile e confrontandolo con tumori umani, lo studio pone le basi per dissezionare i primi stadi della malattia e testare terapie che prendono di mira vie di crescita chiave, come la segnalazione MAPK, prima che questi tumori rari ma gravi si manifestino pienamente.
Citazione: Kommoss, F.K.F., Zhang, J.Y.H., Lynch, B.J. et al. Spatial single cell transcriptomic analysis informs tumor developmental hierarchy of DICER1 syndrome related sarcoma. Nat Commun 17, 4608 (2026). https://doi.org/10.1038/s41467-026-70971-6
Parole chiave: sindrome DICER1, sarcoma, fibroblasti, trascrittomica a singola cellula, modello murino del cancro