Clear Sky Science · de
Räumliche Einzelzell-Transkriptomik zeigt die tumorale Entwicklungshierarchie bei DICER1-Syndrom-assoziierten Sarkomen
Warum diese seltenen Kinderkrebserkrankungen wichtig sind
Das DICER1-Syndrom ist eine seltene vererbte Erkrankung, die das Krebsrisiko erhöht, insbesondere bei Kindern und jungen Erwachsenen. Viele dieser Tumoren sind Sarkome, also Krebsarten, die aus dem Stützgewebe des Körpers entstehen. Obwohl sie in verschiedenen Organen auftreten können, ähneln sie sich mikroskopisch auffallend. Die Studie stellt eine einfache, aber entscheidende Frage: Wo beginnen diese Tumoren, und wie verändern sie sich, während sie wachsen?
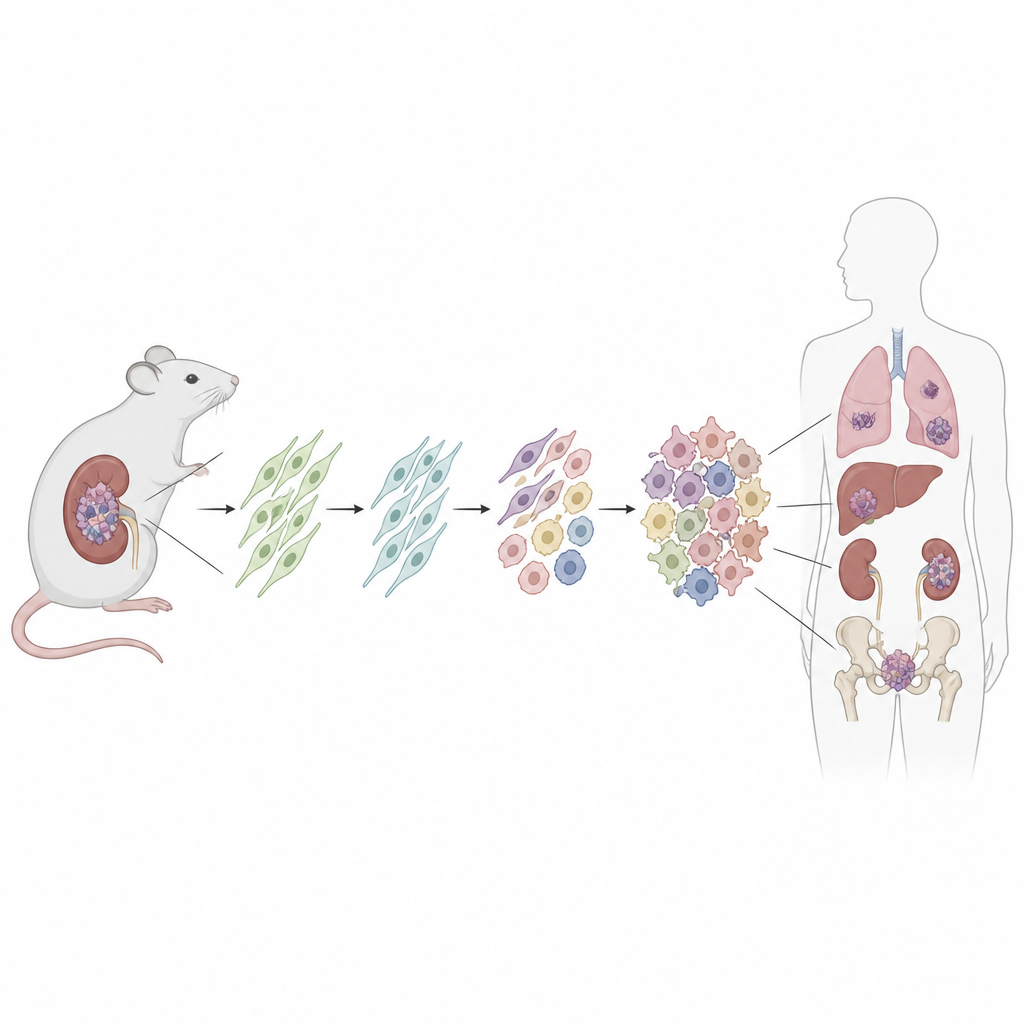
Ein genauerer Blick auf ein krebsprädisponierendes Syndrom
Menschen mit DICER1-Syndrom werden mit einer schädlichen Veränderung in einer Kopie des DICER1-Gens geboren, das an der Verarbeitung winziger RNA-Moleküle beteiligt ist, die viele andere Gene fein regulieren. Tumoren entstehen meist erst, wenn die zweite DICER1-Kopie eine spezifische Fehlfunktion in einer Region namens RNase-IIIb-Domäne entwickelt. Dadurch gerät das normale Gleichgewicht kleiner RNAs und der Genaktivität aus dem Lot. Obwohl die Tumoren in Lunge, Niere, Gehirn und Fortpflanzungsorganen auftreten können, teilen viele ähnliche Strukturen und Verhaltensweisen, was darauf hindeutet, dass sie von einem gemeinsamen Zelltyp ausgehen könnten.
Ein nachvollziehbares Tumormodell in der Maus aufbauen
Um die Ursprungszelle aufzuspüren, erzeugten die Forscher eine Maus, bei der eine patientenähnliche Dicer1-Mutation in einer definierten Gruppe ruhender Stütz-Zellen, den mesenchymalen Stromazellen, eingeschaltet werden kann. Sie verwendeten ein Medikament, um die Mutation nach der Geburt auszulösen, und schalteten gleichzeitig ein fluoreszierendes Etikett ein, das dauerhaft alle Nachkommenzellen markiert. Im Laufe der Zeit entwickelten die meisten dieser Mäuse Nierentumoren, die den menschlichen DICER1-assoziierten Sarkomen sehr ähnelten – von frühen, niedriggradigen Läsionen bis zu großen, aggressiven Massen, die die Niere überwucherten. Die umgebenden Nierenstrukturen, wie etwa die Auskleidung von Zysten, trugen das fluoreszierende Etikett nicht, was zeigt, dass sie selbst nicht tumoral waren.
Ein verwundbarer Fibroblasten-Vorläufer
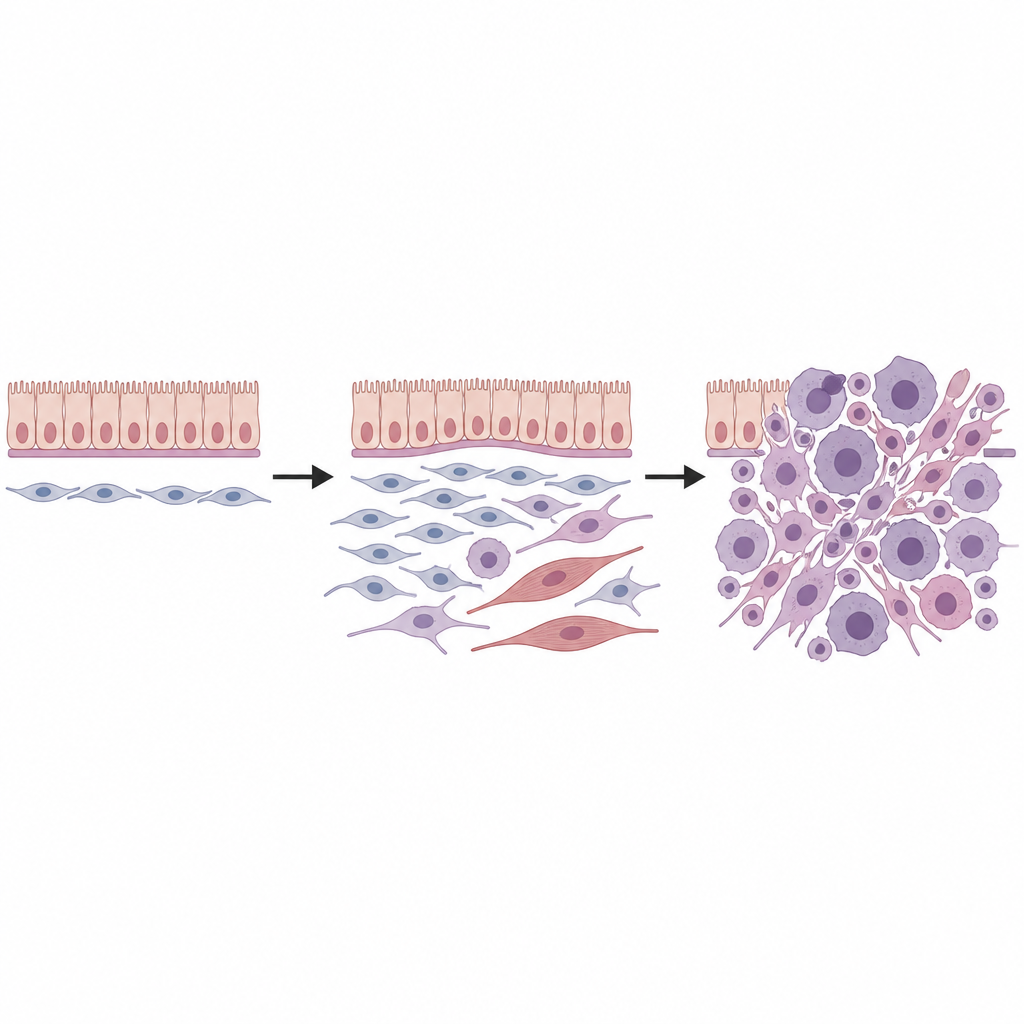
Anschließend kombinierten die Forschenden Einzelzell-RNA-Sequenzierung, die die Genaktivität einzelner Zellen ausliest, mit räumlicher Transkriptomik, die diese Zellen wieder in Gewebeschnitte einordnet. In gesunden Nieren fanden sie mehrere Typen mesenchymaler Zellen, darunter eine spezielle Gruppe von Fibroblasten, die um Blutgefäße, unter Oberflächenepithelien und unter dem Übergangsepithel der Sammelrohre lokalisiert sind. Diese sogenannten universellen Fibroblasten, gekennzeichnet durch eine spezifische Genmischung, standen offenbar an der Wurzel eines kleinen Stammbaums von Stützzellen. Nach Aktivierung der Dicer1-Mutation wurden diese Fibroblasten umprogrammiert: Sie verloren Merkmale reifer Linien und erwarben Eigenschaften eines primitiveren, flexibleren Zustands, der für maligne Veränderungen prädisponiert war.
Die Tumorentwicklung Zelle für Zelle beobachten
Indem sie Tausende einzelner Zellen mittels computergestützter „Pseudotime“-Analyse verfolgten, rekonstruierten die Forscher mehrere Entwicklungswege, die von diesen universellen Fibroblasten ausgehen. Ein Weg führte zu Zellen, die zunächst unreifem Muskel ähnelten und dann voll ausdifferenzierten quergestreiften Muskelzellen glichen, was dem rhabdomyoblastischen Erscheinungsbild vieler DICER1-Sarkome entspricht. Ein anderer Weg brachte fibroblastähnliche Tumorzellen hervor, die chromosomale Gewinne anhäuften, insbesondere auf den Mauschromosomen 1 und 6, und sich dann zu stark proliferierenden Sarkomzellen weiterentwickelten. Zusätzliche genetische Veränderungen, darunter Mutationen bekannter Krebsgene wie p53 und Kras, förderten weiteres Wachstum und aggressives Verhalten, unter anderem durch Abschwächung zellulärer Todesmechanismen und Verstärkung der MAPK-Signalwege.
Maus- und Humantumoren in Verbindung bringen
Um zu prüfen, ob die Ergebnisse beim Menschen relevant sind, wendete das Team räumliche Transkriptomik auf sechzehn menschliche DICER1-assoziierte mesenchymale Tumoren aus verschiedenen Organen und klinischen Subtypen an. Sie entdeckten erneut einen Kernsatz von Zellzuständen: eine Vorläuferpopulation mit dem universellen Fibroblasten-Gen-Signatur, proliferierende Tumorzellen, intermediäre und voll ausdifferenzierte muskelähnliche Zellen sowie eine Umgebung gewöhnlicher Fibroblasten. Die räumliche Anordnung dieser Zellen, einschließlich einer dichten „Kambiumschicht“ von Vorläufern unter epithelialen Auskleidungen, spiegelte die Maus-Tumoren eng wider. Die Überschneidung mit bekannten Zellprogrammen des kindlichen Rhabdomyosarkoms legt nahe, dass zumindest einige dieser muskelähnlichen Tumoren aus Fibroblasten und nicht aus bereits zu Muskelzellen verpflichteten Vorläufern hervorgehen könnten.
Was das für Patientinnen, Patienten und künftige Therapien bedeutet
Die Studie liefert starke Hinweise darauf, dass DICER1-Syndrom-assoziierte Sarkome von einem gemeinsamen Fibroblasten-Vorläufer ausgehen, der normalerweise Gewebe in der Nähe epithelialer Oberflächen und Blutgefäße stützt. Wenn die DICER1-Funktion in diesen Zellen gestört ist, werden sie ungewöhnlich plastisch: Einige richten sich in Richtung einer muskelähnlichen Identität aus, andere entwickeln sich zu schnell wachsenden Sarkomzellen, während sie zusätzliche genetische Schäden anhäufen. Durch den Aufbau eines treuen und rückverfolgbaren Mausmodells und dessen Abgleich mit humanen Tumoren schafft die Arbeit die Grundlage dafür, frühe Krankheitsphasen zu entschlüsseln und Therapien zu testen, die zentrale Wachstumswege wie die MAPK-Signalgebung anvisieren, bevor sich diese seltenen, aber schwerwiegenden Tumoren vollständig manifestieren.
Zitation: Kommoss, F.K.F., Zhang, J.Y.H., Lynch, B.J. et al. Spatial single cell transcriptomic analysis informs tumor developmental hierarchy of DICER1 syndrome related sarcoma. Nat Commun 17, 4608 (2026). https://doi.org/10.1038/s41467-026-70971-6
Schlüsselwörter: DICER1-Syndrom, Sarkom, Fibroblasten, Einzelzell-Transkriptomik, Maus-Krebsmodell