Clear Sky Science · en
Spatial single cell transcriptomic analysis informs tumor developmental hierarchy of DICER1 syndrome related sarcoma
Why these rare childhood cancers matter
DICER1 syndrome is a rare inherited condition that raises the risk of cancers, especially in children and young adults. Many of these tumors are sarcomas, cancers that grow from the body’s supporting tissues. Although they can appear in different organs, they look strikingly alike under the microscope. This study asks a basic but crucial question: where do these cancers start, and how do they change as they grow?
A closer look at a cancer predisposition syndrome
People with DICER1 syndrome are born with a harmful change in one copy of the DICER1 gene, which helps process tiny RNA molecules that fine tune many other genes. Tumors usually develop after the second copy of DICER1 picks up a specific defect in a region called the RNase IIIb domain. This disrupts the normal balance of small RNAs and gene activity. Despite appearing in the lung, kidney, brain, and reproductive organs, many of these tumors share similar structures and behavior, hinting that they may arise from a common type of cell.
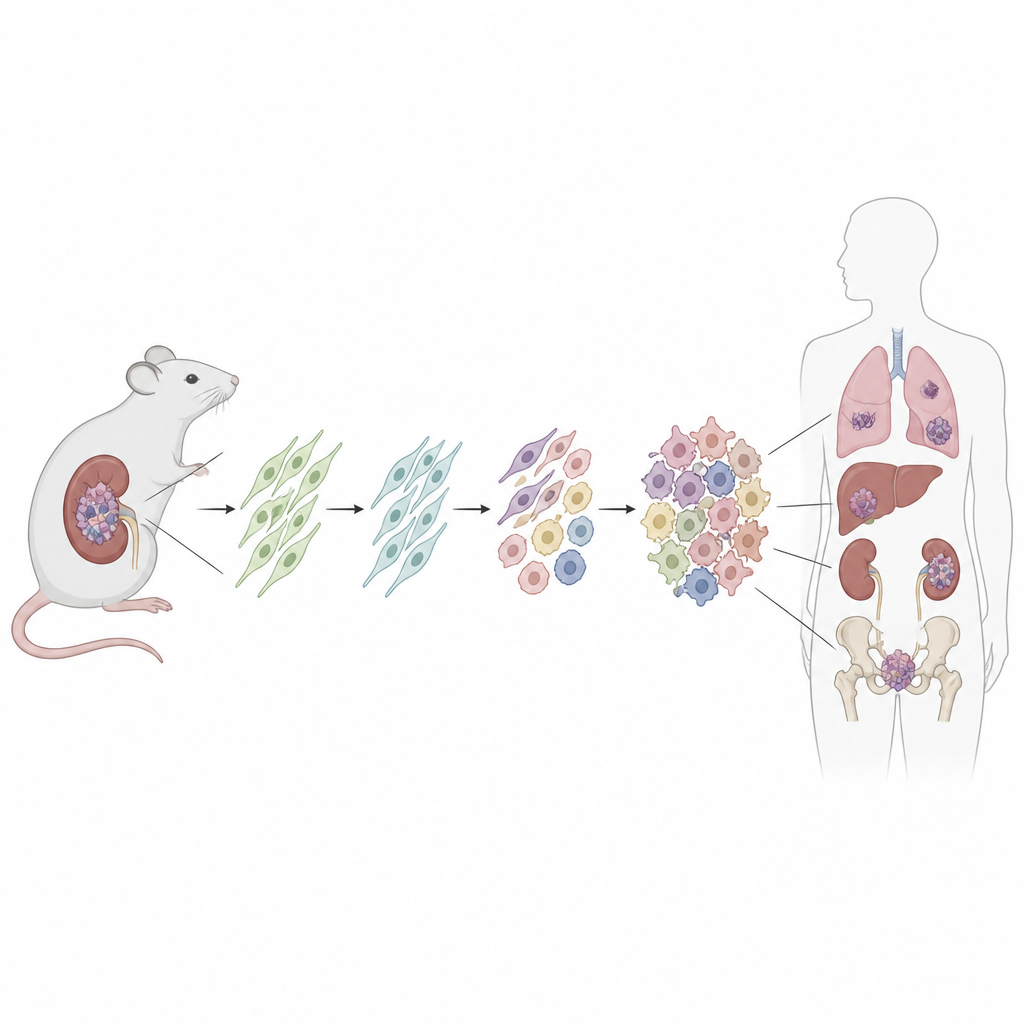
Building a traceable cancer in a mouse
To hunt for the cell of origin, the researchers engineered a mouse in which a patientlike Dicer1 mutation can be switched on in a defined set of quiet support cells called mesenchymal stromal cells. They used a drug to trigger the mutation after birth and also turned on a fluorescent tag to permanently label all descendant cells. Over time, most of these mice developed kidney tumors that closely mirrored human DICER1 related sarcomas, from early low grade lesions to large, aggressive masses that overgrew the kidney. The surrounding kidney structures, such as the lining of cysts, did not carry the fluorescent tag, showing they were not themselves cancerous.
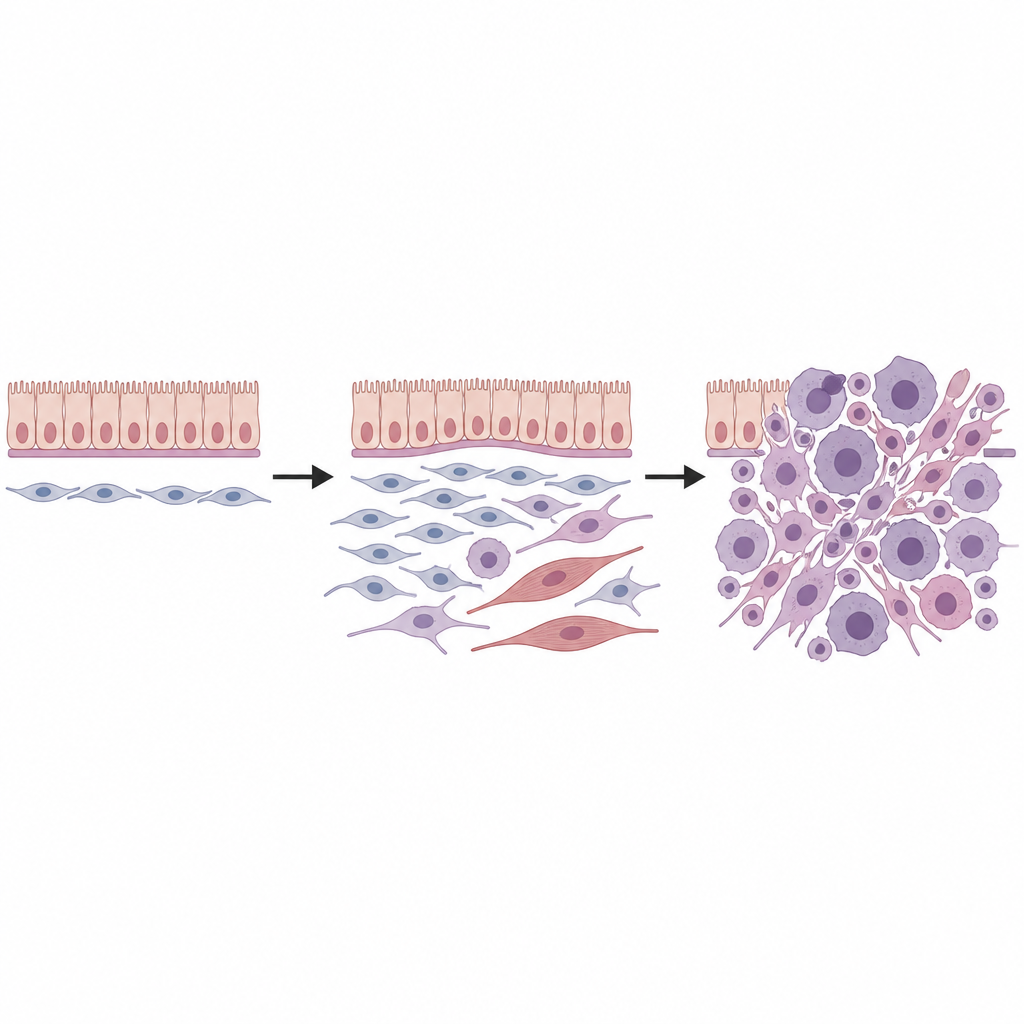
Finding a vulnerable fibroblast ancestor
Next, the team combined single cell RNA sequencing, which reads out gene activity cell by cell, with spatial transcriptomics, which maps those cells back into tissue sections. In healthy kidneys, they found several types of mesenchymal cells, including a special group of fibroblasts located around blood vessels, under surface linings, and beneath the transitional epithelium of collecting ducts. These so called universal fibroblasts, marked by a specific mix of genes, appeared to sit at the root of a small family tree of support cells. When the Dicer1 mutation was activated, these fibroblasts were reprogrammed: they lost markers of mature lineages and gained features of a more primitive, flexible state that was poised for malignant change.
Watching tumors evolve cell by cell
By following thousands of individual cells through computational “pseudotime” analysis, the researchers reconstructed several developmental paths starting from these universal fibroblasts. One route led toward cells that began to resemble immature muscle and then fully formed skeletal muscle like cells, mirroring the rhabdomyoblastic appearance seen in many DICER1 sarcomas. Another route produced fibroblastlike tumor cells that accumulated chromosomal gains, particularly involving mouse chromosomes 1 and 6, and then progressed into highly proliferative sarcoma cells. Additional genetic hits, including mutations in classic cancer genes such as p53 and Kras, further fueled growth and aggressive behavior, in part by dialing down cell death safeguards and ramping up MAPK signaling pathways.
Connecting mouse and human tumors
To test whether these findings were relevant to people, the team applied spatial transcriptomics to sixteen human DICER1 related mesenchymal tumors from various organs and clinical subtypes. They again discovered a core set of cell states: a progenitor population sharing the universal fibroblast gene signature, proliferating tumor cells, intermediate and fully musclelike cells, and a background of more ordinary fibroblasts. The spatial arrangement of these cells, including a dense “cambium layer” of progenitors beneath epithelial linings, closely echoed the mouse tumors. The overlap with known cell programs in childhood rhabdomyosarcoma suggests that at least some of these musclelike cancers may spring from fibroblasts rather than from committed muscle precursors.
What this means for patients and future treatments
This work provides strong evidence that DICER1 syndrome related sarcomas arise from a common fibroblast ancestor that normally supports tissues near epithelial surfaces and blood vessels. When DICER1 function is disturbed in these cells, they become unusually plastic: some move toward a musclelike identity, while others evolve into fast growing sarcoma cells as they collect further genetic damage. By building a faithful and traceable mouse model and aligning it with human tumors, the study lays the groundwork for dissecting early disease steps and testing therapies that target key growth pathways, such as MAPK signaling, before these rare but serious cancers fully take hold.
Citation: Kommoss, F.K.F., Zhang, J.Y.H., Lynch, B.J. et al. Spatial single cell transcriptomic analysis informs tumor developmental hierarchy of DICER1 syndrome related sarcoma. Nat Commun 17, 4608 (2026). https://doi.org/10.1038/s41467-026-70971-6
Keywords: DICER1 syndrome, sarcoma, fibroblasts, single cell transcriptomics, mouse cancer model