Clear Sky Science · fr
Analyse transcriptomique spatiale unicellulaire éclaire la hiérarchie développementale des tumeurs dans les sarcomes liés au syndrome DICER1
Pourquoi ces cancers infantiles rares comptent
Le syndrome DICER1 est une affection héréditaire rare qui augmente le risque de cancers, en particulier chez les enfants et les jeunes adultes. Nombre de ces tumeurs sont des sarcomes, des cancers issus des tissus de soutien de l’organisme. Bien qu’ils puissent apparaître dans différents organes, ils se ressemblent remarquablement au microscope. Cette étude pose une question simple mais cruciale : où ces cancers débutent-ils et comment évoluent-ils en grandissant ?
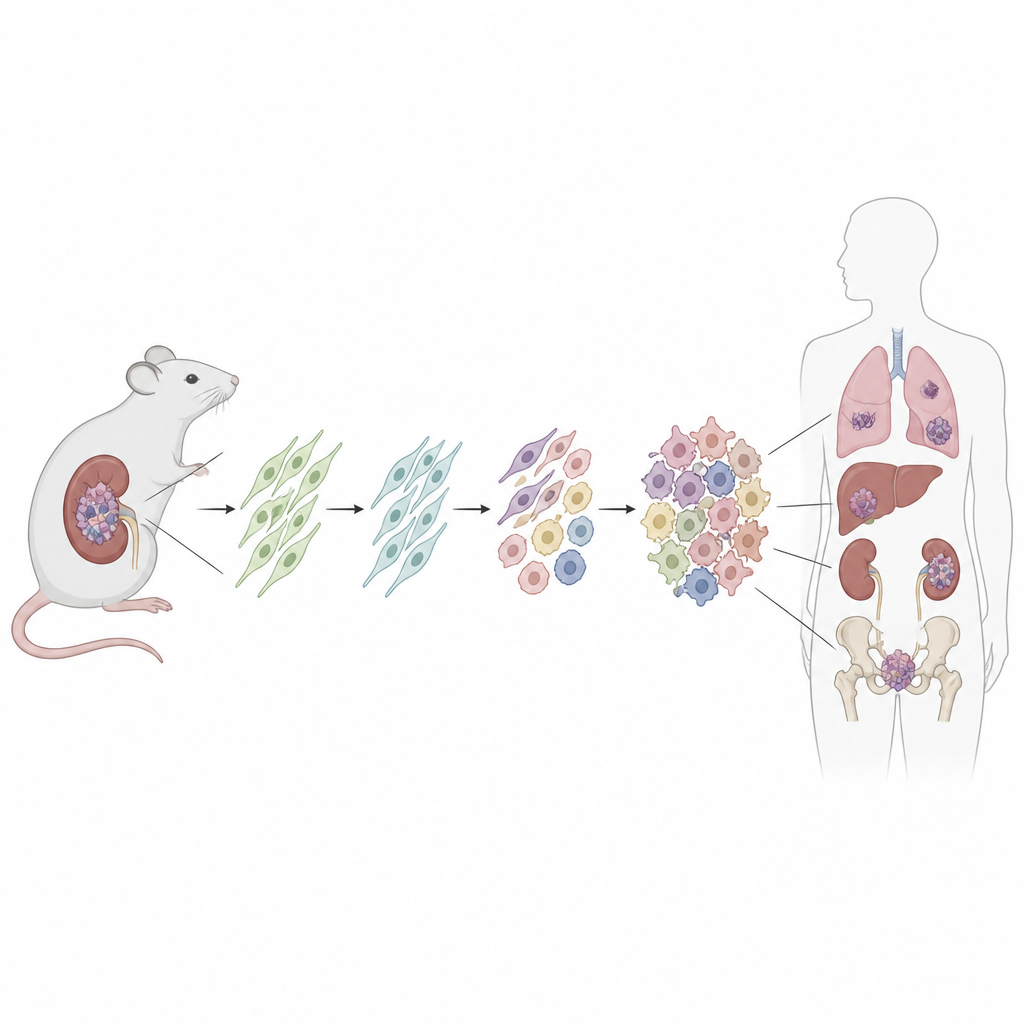
Un examen approfondi d’un syndrome de prédisposition au cancer
Les personnes atteintes du syndrome DICER1 naissent avec une altération délétère d’une copie du gène DICER1, qui participe au traitement de petits ARN régulateurs modulant de nombreux autres gènes. Les tumeurs se développent généralement après qu’une seconde copie de DICER1 ait acquis une anomalie spécifique dans une région appelée domaine RNase IIIb. Cela perturbe l’équilibre normal des petits ARN et de l’activité génique. Malgré leur apparition dans le poumon, le rein, le cerveau et les organes reproducteurs, beaucoup de ces tumeurs partagent des structures et des comportements similaires, ce qui suggère qu’elles pourraient provenir d’un même type cellulaire.
Construire un cancer traçable chez la souris
Pour rechercher la cellule d’origine, les chercheurs ont conçu une souris dans laquelle une mutation de Dicer1 de type patient peut être activée dans un ensemble défini de cellules de soutien peu actives appelées cellules stromales mésenchymateuses. Ils ont utilisé un médicament pour déclencher la mutation après la naissance et ont aussi activé un marqueur fluorescent afin d’étiqueter de façon permanente toutes les cellules descendantes. Au fil du temps, la plupart de ces souris ont développé des tumeurs rénales reproduisant fidèlement les sarcomes liés à DICER1 chez l’humain, depuis des lésions précoces de bas grade jusqu’à de grosses masses agressives envahissant le rein. Les structures rénales environnantes, comme l’épithélium des kystes, ne portaient pas le marqueur fluorescent, montrant qu’elles n’étaient pas elles‑mêmes tumorales.
Identification d’un ancêtre fibroblastique vulnérable
Ensuite, l’équipe a combiné le séquençage ARN unicellulaire, qui lit l’activité génique cellule par cellule, avec la transcriptomique spatiale, qui remet ces cellules dans leur contexte tissulaire. Dans des reins sains, ils ont trouvé plusieurs types de cellules mésenchymateuses, dont un groupe particulier de fibroblastes situé autour des vaisseaux, sous les revêtements de surface et sous l’épithélium de transition des conduits collecteurs. Ces fibroblastes dits universels, marqués par un mélange spécifique de gènes, semblaient occuper la racine d’un petit arbre généalogique de cellules de soutien. Lors de l’activation de la mutation Dicer1, ces fibroblastes ont été reprogrammés : ils ont perdu les marqueurs des lignées matures et acquis des caractéristiques d’un état plus primitif et plastique, prêt à une transformation maligne.
Observer l’évolution des tumeurs cellule par cellule
En suivant des milliers de cellules individuelles grâce à une analyse computationnelle en « pseudotemps », les chercheurs ont reconstruit plusieurs trajectoires développementales partant de ces fibroblastes universels. Une voie a mené à des cellules commençant à ressembler à des cellules musculaires immatures puis à des cellules de type muscle squelettique pleinement formées, reflétant l’apparence rhabdomyoblastique observée dans de nombreux sarcomes DICER1. Une autre voie a produit des cellules tumorales de type fibroblastique qui ont accumulé des gains chromosomiques, notamment sur les chromosomes murins 1 et 6, puis ont progressé vers des cellules sarcomateuses fortement prolifératives. Des coups génétiques supplémentaires, incluant des mutations dans des gènes classiques du cancer comme p53 et Kras, ont alimenté la croissance et l’agressivité, en partie en réduisant les garde‑fous de la mort cellulaire et en amplifiant les voies de signalisation MAPK.
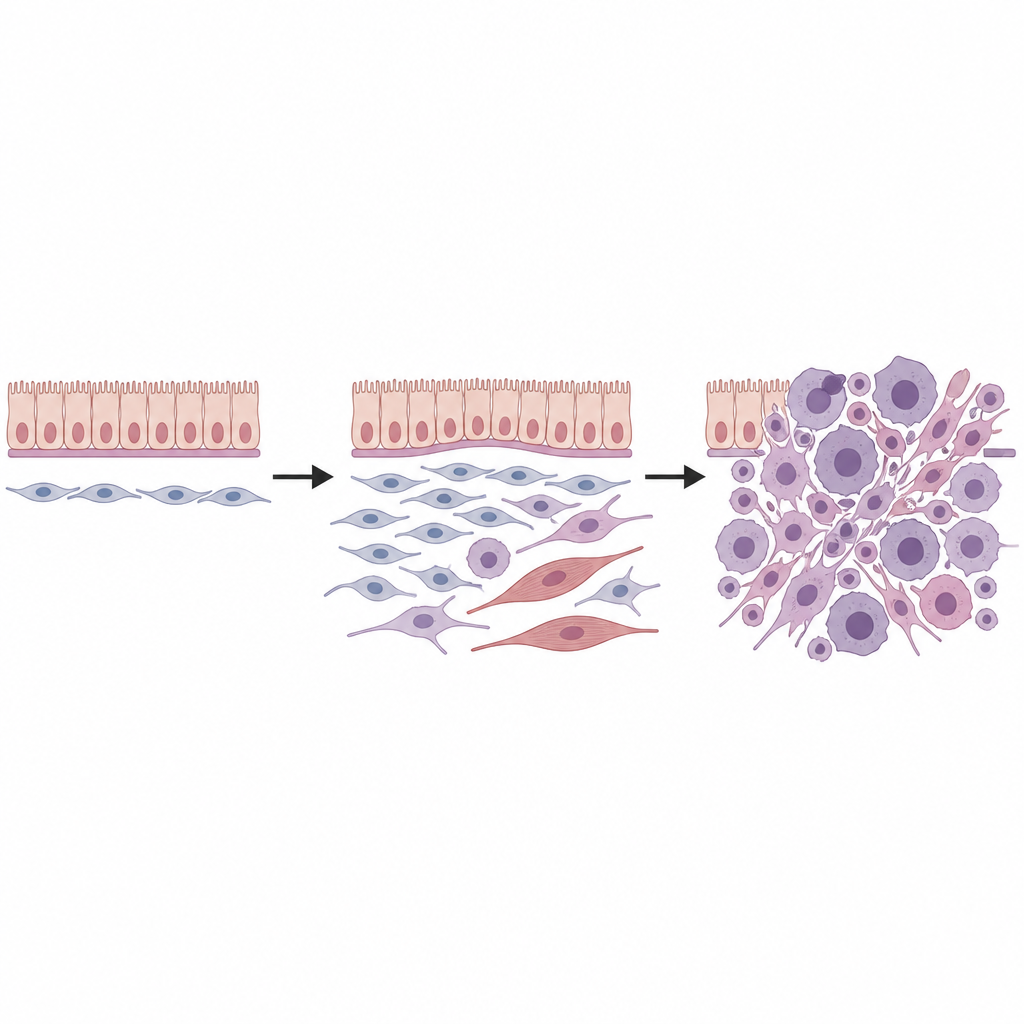
Relier les tumeurs murines et humaines
Pour vérifier la pertinence chez l’humain, l’équipe a appliqué la transcriptomique spatiale à seize tumeurs mésenchymateuses humaines liées à DICER1, issues de divers organes et sous‑types cliniques. Ils ont de nouveau identifié un ensemble central d’états cellulaires : une population de progéniteurs partageant la signature génique des fibroblastes universels, des cellules tumorales proliférantes, des cellules intermédiaires et pleinement de type musculaire, et un fond de fibroblastes plus ordinaires. L’agencement spatial de ces cellules, incluant une dense « couche cambiale » de progéniteurs sous les revêtements épithéliaux, reflétait étroitement les tumeurs murines. Le chevauchement avec des programmes cellulaires connus du rhabdomyosarcome pédiatrique suggère qu’au moins certains de ces cancers de type musculaire peuvent dériver de fibroblastes plutôt que de précurseurs musculaires engagés.
Ce que cela signifie pour les patients et les traitements futurs
Ce travail apporte des preuves solides que les sarcomes liés au syndrome DICER1 proviennent d’un ancêtre fibroblastique commun qui soutient normalement les tissus proches des surfaces épithéliales et des vaisseaux. Lorsque la fonction de DICER1 est perturbée dans ces cellules, elles deviennent exceptionnellement plastiques : certaines évoluent vers une identité de type musculaire, tandis que d’autres se transforment en cellules sarcomateuses à croissance rapide à mesure qu’elles accumulent d’autres altérations génétiques. En construisant un modèle murin fidèle et traçable et en l’alignant sur des tumeurs humaines, l’étude pose les bases pour disséquer les étapes précoces de la maladie et tester des thérapies ciblant des voies de croissance clés, comme la signalisation MAPK, avant que ces cancers rares mais graves ne se développent pleinement.
Citation: Kommoss, F.K.F., Zhang, J.Y.H., Lynch, B.J. et al. Spatial single cell transcriptomic analysis informs tumor developmental hierarchy of DICER1 syndrome related sarcoma. Nat Commun 17, 4608 (2026). https://doi.org/10.1038/s41467-026-70971-6
Mots-clés: syndrome DICER1, sarcome, fibroblastes, transcriptomique unicellulaire, modèle murin de cancer