Clear Sky Science · es
Análisis transcriptómico espacial unicelular informa sobre la jerarquía de desarrollo tumoral en el sarcoma relacionado con el síndrome DICER1
Por qué importan estos raros cánceres infantiles
El síndrome DICER1 es una enfermedad hereditaria poco frecuente que aumenta el riesgo de desarrollar cánceres, sobre todo en niños y adultos jóvenes. Muchos de estos tumores son sarcomas, neoplasias que se originan en los tejidos de sostén del organismo. Aunque pueden aparecer en distintos órganos, se parecen de forma llamativa al observarlos al microscopio. Este estudio plantea una pregunta básica pero crucial: ¿dónde se inician estos cánceres y cómo cambian a medida que crecen?
Una mirada más detallada a un síndrome de predisposición al cáncer
Las personas con síndrome DICER1 nacen con una alteración dañina en una copia del gen DICER1, que participa en el procesamiento de pequeñas moléculas de ARN que regulan con precisión muchos otros genes. Los tumores suelen desarrollarse cuando la segunda copia de DICER1 adquiere un defecto específico en una región llamada dominio RNasa IIIb. Esto altera el equilibrio normal de los pequeños ARN y la actividad génica. A pesar de aparecer en pulmón, riñón, cerebro y órganos reproductores, muchos de estos tumores comparten estructuras y comportamientos similares, lo que sugiere que podrían originarse a partir de un tipo celular común.
Construyendo un cáncer rastreable en ratón
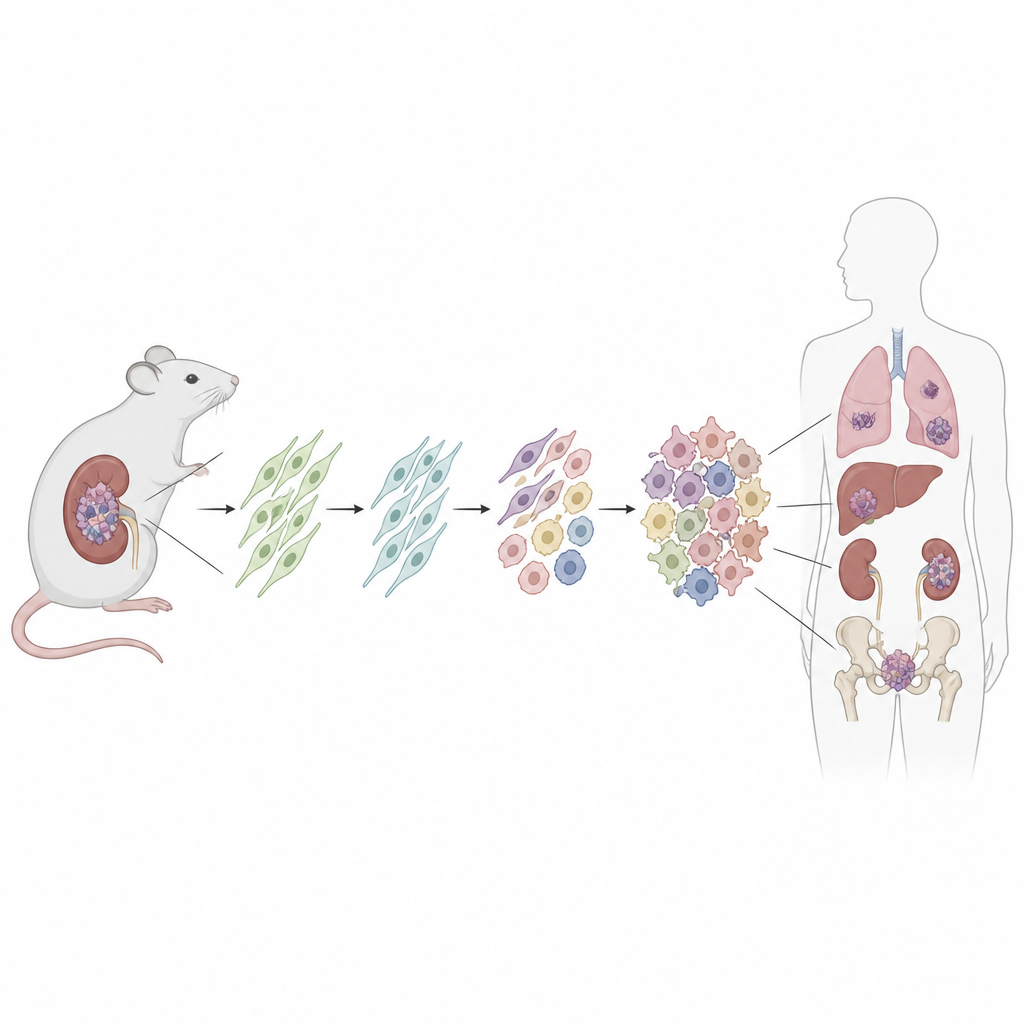
Para buscar la célula de origen, los investigadores diseñaron un ratón en el que una mutación en Dicer1 similar a la humana puede activarse en un conjunto definido de células de soporte en reposo denominadas células estromales mesenquimales. Usaron un fármaco para desencadenar la mutación tras el nacimiento y también activaron una etiqueta fluorescente para marcar de forma permanente a todas las células descendientes. Con el tiempo, la mayoría de estos ratones desarrollaron tumores renales que reproducían fielmente los sarcomas humanos relacionados con DICER1, desde lesiones tempranas de bajo grado hasta masas grandes y agresivas que invadían el riñón. Las estructuras renales circundantes, como el revestimiento de quistes, no portaban la etiqueta fluorescente, lo que indica que no eran cancerosas.
Encontrando un ancestro fibroblástico vulnerable
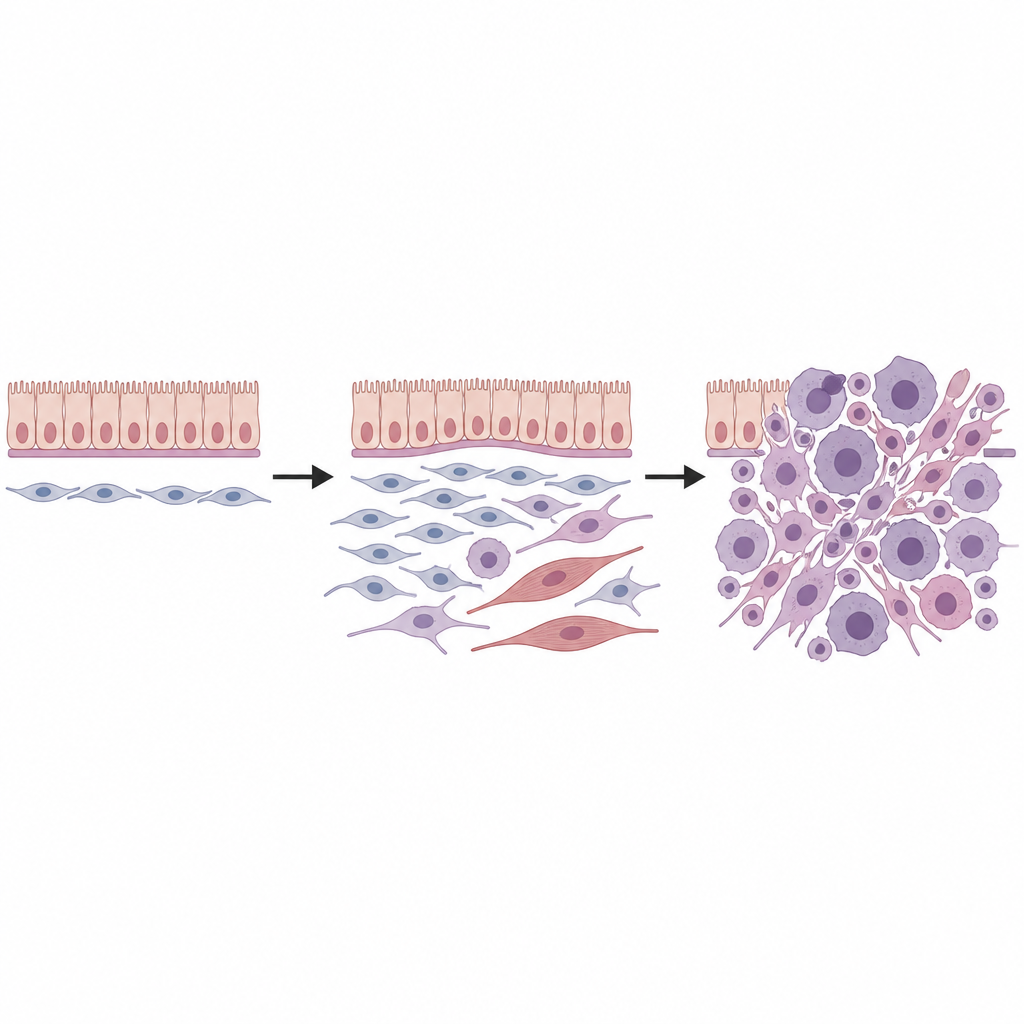
A continuación, el equipo combinó secuenciación de ARN unicelular, que registra la actividad génica célula por célula, con transcriptómica espacial, que sitúa esas células de nuevo en cortes de tejido. En riñones sanos hallaron varios tipos de células mesenquimales, incluida un grupo especial de fibroblastos ubicados alrededor de vasos sanguíneos, bajo revestimientos superficiales y bajo el epitelio transicional de los conductos colectores. Estos llamados fibroblastos universales, marcados por una combinación específica de genes, parecían ocupar la raíz de un pequeño árbol genealógico de células de sostén. Cuando se activó la mutación de Dicer1, estos fibroblastos fueron reprogramados: perdieron marcadores de linajes maduros y adquirieron rasgos de un estado más primitivo y flexible predispuesto al cambio maligno.
Observando la evolución tumoral célula por célula
Siguiendo a miles de células individuales mediante análisis computacionales de "pseudotiempo", los investigadores reconstruyeron varias trayectorias de desarrollo que partían de estos fibroblastos universales. Una ruta avanzó hacia células que empezaron a parecerse a músculo inmaduro y luego a células semejantes al músculo esquelético maduro, reflejando la apariencia rabdomioblástica observada en muchos sarcomas DICER1. Otra ruta dio lugar a células tumorales con aspecto fibroblástico que acumularon ganancias cromosómicas, en particular en los cromosomas murinos 1 y 6, y luego progresaron hacia células sarcomatosas altamente proliferativas. Golpes genéticos adicionales, incluidas mutaciones en genes clásicos del cáncer como p53 y Kras, impulsaron aún más el crecimiento y el comportamiento agresivo, en parte al reducir mecanismos de muerte celular y potenciar las vías de señalización MAPK.
Conectando tumores de ratón y humanos
Para comprobar si estos hallazgos eran relevantes en humanos, el equipo aplicó transcriptómica espacial a dieciséis tumores mesenquimales humanos relacionados con DICER1 procedentes de diversos órganos y subtipos clínicos. De nuevo descubrieron un conjunto central de estados celulares: una población progenitora que comparte la firma génica de los fibroblastos universales, células tumorales proliferantes, células intermedias y totalmente semejantes al músculo, y un fondo de fibroblastos más ordinarios. La disposición espacial de estas células, incluida una densa "capa cambial" de progenitores bajo revestimientos epiteliales, reflejaba de cerca los tumores de ratón. La superposición con programas celulares conocidos en el rabdomiosarcoma infantil sugiere que al menos algunos de estos cánceres con aspecto muscular podrían surgir de fibroblastos en lugar de precursores musculares comprometidos.
Qué significa esto para los pacientes y tratamientos futuros
Este trabajo aporta pruebas sólidas de que los sarcomas relacionados con el síndrome DICER1 se originan en un ancestro fibroblástico común que normalmente sostiene tejidos cercanos a superficies epiteliales y vasos sanguíneos. Cuando la función de DICER1 se altera en estas células, se vuelven extraordinariamente plásticas: algunas derivan hacia una identidad musculosa, mientras que otras evolucionan a células sarcomatosas de rápido crecimiento a medida que acumulan más daño genético. Al construir un modelo murino fiel y rastreable y alinearlo con tumores humanos, el estudio sienta las bases para diseccionar los pasos iniciales de la enfermedad y probar terapias que apunten a vías de crecimiento clave, como la señalización MAPK, antes de que estos cánceres raros pero graves se instauren por completo.
Cita: Kommoss, F.K.F., Zhang, J.Y.H., Lynch, B.J. et al. Spatial single cell transcriptomic analysis informs tumor developmental hierarchy of DICER1 syndrome related sarcoma. Nat Commun 17, 4608 (2026). https://doi.org/10.1038/s41467-026-70971-6
Palabras clave: Síndrome DICER1, sarcoma, fibroblastos, transcriptómica unicelular, modelo murino de cáncer