Clear Sky Science · pt
Descoberta in silico de análogos tioglicosídicos como inibidores do sítio doador da glicosiltransferase LgtC
Por que enfraquecer os germes pode ser melhor do que matá‑los
Hospitais ao redor do mundo enfrentam infecções causadas por bactérias que já não respondem a muitos antibióticos. Em vez de tentar matar esses microrganismos diretamente, alguns cientistas exploram uma tática diferente: desarmá‑los discretamente para que nosso sistema imunológico e os medicamentos existentes façam o resto. Este estudo usa modelos computacionais para buscar novas pequenas moléculas, chamadas tioglicosídeos, que poderiam bloquear uma enzima bacteriana chave e tornar patógenos Gram‑negativos perigosos muito menos capazes de causar doença.
O problema das defesas externas resistentes
Muitas das bactérias mais problemáticas, como espécies de Neisseria e outros germes Gram‑negativos, estão revestidas por uma capa externa complexa de moléculas açucaradas. Parte dessa cobertura, conhecida como lipooligosacarídeo, ajuda a evitar nossas defesas imunes e a resistir a antibióticos. Construir essa armadura exige um conjunto de enzimas especializadas. Uma delas, chamada LgtC, conecta uma unidade de açúcar chamada galactose à camada externa em formação. Se a LgtC for bloqueada, a estrutura de superfície fica incompleta e as bactérias ficam mais vulneráveis. Como células humanas não usam LgtC, ela é um alvo atraente para fármacos que possam incapacitar patógenos sem nos prejudicar.
Usando computadores para vasculhar o espaço químico
Os pesquisadores concentraram‑se em uma família de moléculas semelhantes a açúcares chamadas tioglicosídeos, que têm um átomo de enxofre no lugar de uma ligação de oxigênio encontrada em açúcares naturais. Trabalhos anteriores mostraram que dois desses compostos, chamados FucSBn e BacSBn, podem interferir na montagem dos açúcares bacterianos. Aqui, a equipe pesquisou o grande banco de dados PubChem em busca de moléculas que se assemelhassem de perto a esses dois “isquiadores metabólicos”. Mantiveram apenas candidatos que, com base em regras padronizadas de semelhança a fármacos e perfis previstos de absorção e segurança, pareciam adequados para desenvolvimento como medicamentos. Essa etapa de filtração gerou 18 análogos promissores que eram pequenos o suficiente, não excessivamente lipofílicos nem hidrofílicos e pouco prováveis de ser altamente tóxicos em doses terapêuticas.
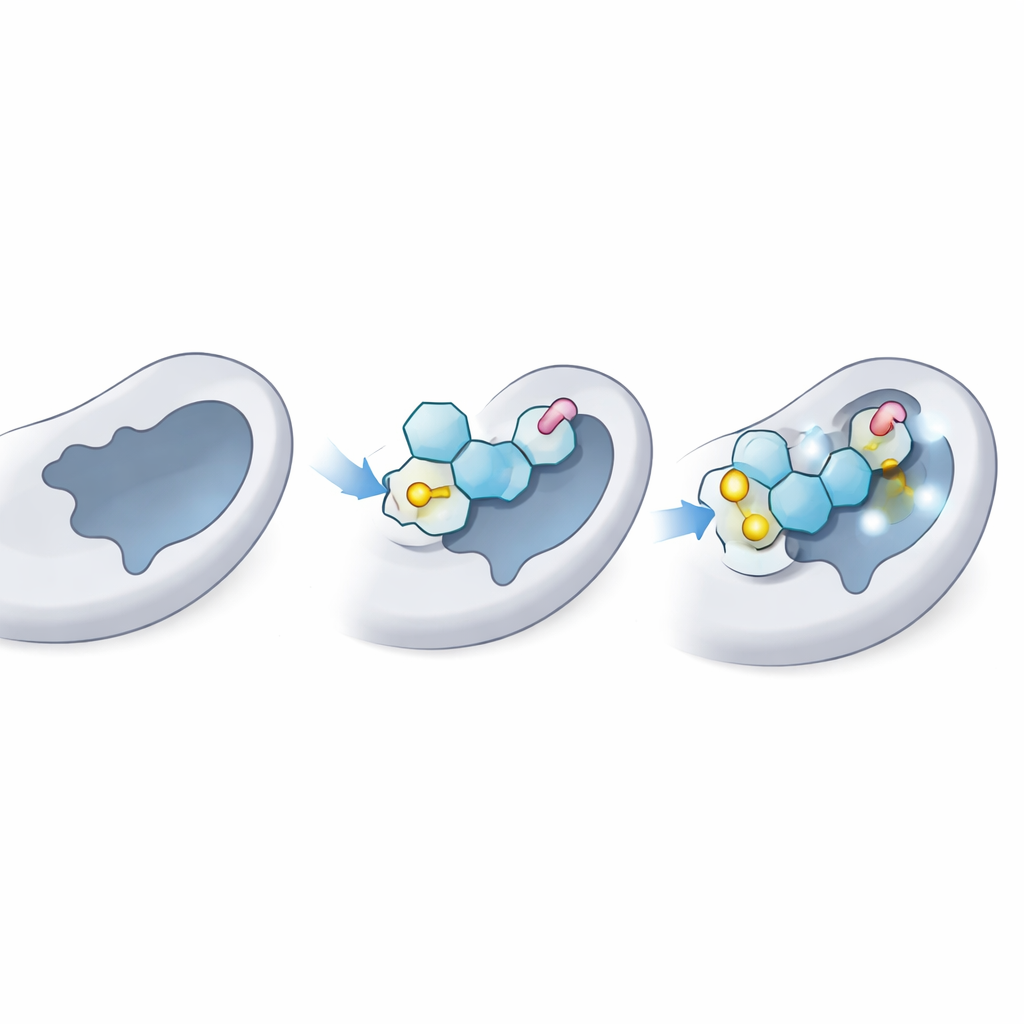
Testando o quão bem os candidatos se ajustam à enzima
Em seguida, os cientistas usaram docking molecular, uma espécie de teste virtual de fechadura e chave, para ver quão firmemente cada tioglicosídeo poderia se acomodar no sítio doador da LgtC — o bolso onde o doador de açúcar natural normalmente se liga. Primeiro confirmaram que seu método conseguia “redockar” corretamente o açúcar doador natural na estrutura 3D conhecida da LgtC, correspondendo aos dados experimentais. Em seguida, dockaram os 18 análogos milhares de vezes. Vários, especialmente três rotulados C‑5, C‑14 e C‑18, mostraram consistentemente previsão de ligação mais forte do que o doador natural, sugerindo que poderiam competir efetivamente pelo mesmo ponto da enzima.
Observando a interação em movimento
O docking oferece uma imagem estática; a equipe então executou simulações de dinâmica molecular de 100 nanossegundos para ver como a enzima e cada ligante se comportavam ao longo do tempo em um ambiente aquoso virtual. Essas simulações acompanham o quanto o complexo oscila, quão compacto a proteína permanece e quais contatos persistem. Os melhores tioglicosídeos mantiveram uma pose estável no bolso doador, com movimento apenas moderado, comparável ao do açúcar natural. Eles preservaram ligações de hidrogênio chave e contatos de curto alcance com os mesmos resíduos âncora que normalmente seguram o doador real no lugar, além de adicionar interações estabilizadoras extras graças às suas ligações de enxofre e “caudas” aromáticas. A forma e a flexibilidade globais da proteína permaneceram dentro de limites saudáveis, indicando que a enzima não estava sendo distorcida, apenas bloqueada.
O que isso significa para tratamentos futuros
Em conjunto, a triagem virtual, as pontuações de docking e as longas simulações apontam para um pequeno conjunto de molduras tioglicosídicas — especialmente C‑5, C‑14 e C‑18 — como fortes candidatas a atuar como bloqueadoras competitivas da LgtC. Em termos simples, essas moléculas parecem capazes de ocupar o sítio de ligação ao açúcar da enzima por tempo suficiente e com afinidade suficiente para impedir que os blocos construtores reais entrem. Isso deve atrapalhar a construção da cobertura protetora das bactérias, enfraquecendo os microrganismos sem necessariamente matá‑los. O trabalho é inteiramente computacional e ainda precisa ser confirmado em experimentos com enzimas e células, mas oferece um roteiro claro para químicos: esses tioglicosídeos fornecem plantas‑base iniciais para projetar uma nova geração de fármacos antivirulência que poderiam ajudar a conter infecções Gram‑negativas multirresistentes.
Citação: Sierra-Hernández, O., Saurith-Coronell, O., Alcázar, J.J. et al. In silico discovery of thioglycoside analogues as donor-site inhibitors of glycosyltransferase LgtC. Sci Rep 16, 13807 (2026). https://doi.org/10.1038/s41598-026-43638-x
Palavras-chave: antivirulência, bactérias Gram-negativas, glicosiltransferase LgtC, inibidores tioglicosídicos, descoberta de fármacos in silico