Clear Sky Science · fr
Découverte in silico d’analogues thioglycosidiques comme inhibiteurs du site donneur de la glycosyltransférase LgtC
Pourquoi affaiblir les germes peut être préférable que les tuer
Les hôpitaux du monde entier sont confrontés à des infections provoquées par des bactéries qui ne répondent plus à de nombreux antibiotiques. Plutôt que d’essayer d’éliminer ces microbes de manière directe, certains scientifiques explorent une stratégie différente : les désarmer discrètement pour que notre système immunitaire et les médicaments existants fassent le reste. Cette étude utilise des modèles informatiques pour rechercher de nouvelles petites molécules, appelées thioglycosides, susceptibles de bloquer une enzyme bactérienne clé et de rendre des agents pathogènes à Gram négatif beaucoup moins capables de provoquer des maladies.
Le problème des remparts externes résistants
Beaucoup des bactéries les plus préoccupantes, comme les espèces de Neisseria et d’autres germes à Gram négatif, sont enveloppées d’une couche externe complexe de molécules sucrées. Une partie de cette couche, connue sous le nom de lipooligosaccharide, les aide à échapper à nos défenses immunitaires et à résister aux antibiotiques. La construction de ce rempart nécessite un ensemble d’enzymes spécialisées. L’une d’elles, nommée LgtC, fixe un sucre appelé galactose à la couche externe en formation. Si LgtC est bloquée, la structure de surface reste incomplète et les bactéries deviennent plus vulnérables. Comme les cellules humaines n’utilisent pas LgtC, cette enzyme constitue une cible attrayante pour des médicaments susceptibles d’affaiblir les pathogènes sans nous nuire.
Utiliser l’informatique pour sonder l’espace chimique
Les chercheurs se sont concentrés sur une famille de molécules de type sucre appelées thioglycosides, qui comportent un atome de soufre à la place d’un lien oxygène présent dans les sucres naturels. Des travaux antérieurs ont montré que deux de ces composés, nommés FucSBn et BacSBn, peuvent perturber l’assemblage des sucres bactériens. Ici, l’équipe a exploré la vaste base de données PubChem à la recherche de molécules ressemblant étroitement à ces deux « leurres métaboliques ». Ils n’ont conservé que les candidats qui, d’après les règles usuelles de « drug‑likeness » et les profils prédits d’absorption et de sécurité, semblaient adaptés au développement pharmaceutique. Cette étape de filtrage a donné 18 analogues prometteurs, suffisamment petits, ni trop lipophiles ni trop hydrophiles, et peu susceptibles d’être hautement toxiques à des doses thérapeutiques.
Tester l’ajustement des candidats dans l’enzyme
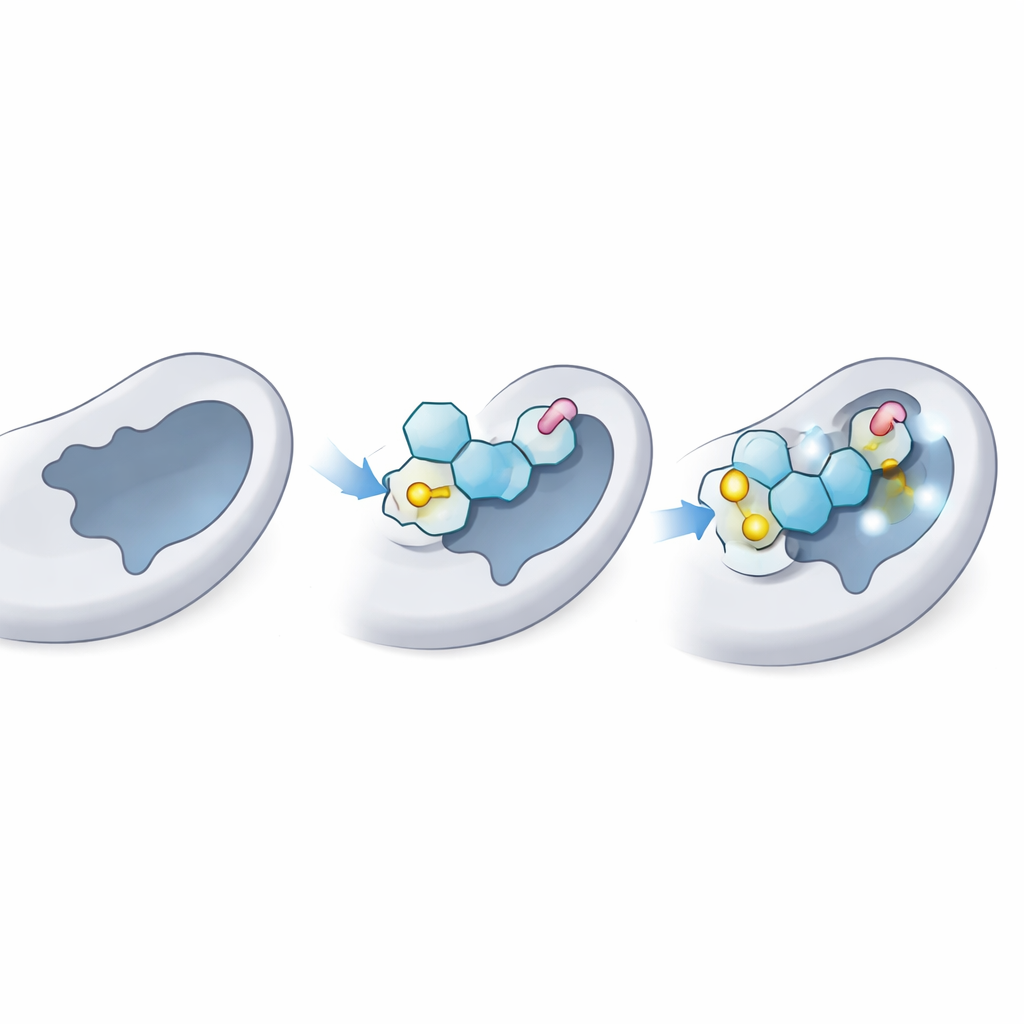
Ensuite, les scientifiques ont utilisé le docking moléculaire, une sorte de test virtuel de serrure et clé, pour évaluer la façon dont chaque thioglycoside pourrait s’insérer dans le site donneur de LgtC — la poche où se lie normalement le donneur de sucre naturel. Ils ont d’abord confirmé que leur méthode pouvait correctement « redocker » le sucre donneur naturel dans la structure 3D connue de LgtC, en accord avec les données expérimentales. Puis ils ont docké les 18 analogues des milliers de fois. Plusieurs d’entre eux, en particulier trois désignés C‑5, C‑14 et C‑18, ont montré de façon constante une affinité prédite supérieure à celle du donneur naturel, ce qui suggère qu’ils pourraient efficacement concurrencer ce dernier pour le même site sur l’enzyme.
Observer l’interaction en mouvement
Le docking fournit une image statique ; l’équipe a ensuite exécuté des simulations de dynamique moléculaire de 100 nanosecondes pour voir comment l’enzyme et chaque ligand se comportaient au fil du temps dans un environnement aqueux virtuel. Ces simulations suivent l’amplitude des fluctuations du complexe, la compacité de la protéine et les contacts qui persistent. Les meilleurs thioglycosides ont conservé une pose stable dans la poche donneuse, avec seulement des mouvements modestes comparables à ceux du sucre naturel. Ils ont maintenu des liaisons hydrogène clés et des contacts à courte distance avec les mêmes résidus d’ancrage qui retiennent normalement le donneur réel, tout en ajoutant des interactions stabilisantes supplémentaires grâce à leurs liaisons soufrées et à leurs « queues » aromatiques. La forme globale et la flexibilité de la protéine sont restées dans des limites saines, indiquant que l’enzyme n’était pas déformée, mais simplement bloquée.
Ce que cela signifie pour les traitements futurs
Pris ensemble, le criblage virtuel, les scores de docking et les longues simulations désignent un petit ensemble d’échafaudages thioglycosidiques — en particulier C‑5, C‑14 et C‑18 — comme de solides candidats pour agir comme bloqueurs compétitifs de LgtC. En termes simples, ces molécules semblent capables d’occuper le site de liaison au sucre de l’enzyme suffisamment longtemps et avec assez d’affinité pour empêcher l’entrée des vrais éléments constructeurs. Cela devrait perturber la construction du revêtement protecteur des bactéries, affaiblissant les microbes sans nécessairement les tuer. Le travail est entièrement computationnel et doit encore être confirmé par des expériences enzymatiques et cellulaires, mais il offre une feuille de route claire pour les chimistes : ces thioglycosides fournissent des plans de départ pour concevoir la prochaine génération de médicaments antivirulence susceptibles d’aider à maîtriser les infections multirésistantes à Gram négatif.
Citation: Sierra-Hernández, O., Saurith-Coronell, O., Alcázar, J.J. et al. In silico discovery of thioglycoside analogues as donor-site inhibitors of glycosyltransferase LgtC. Sci Rep 16, 13807 (2026). https://doi.org/10.1038/s41598-026-43638-x
Mots-clés: antivirulence, bactéries à Gram négatif, glycosyltransférase LgtC, inhibiteurs thioglycosidiques, découverte de médicaments in silico