Clear Sky Science · en
In silico discovery of thioglycoside analogues as donor-site inhibitors of glycosyltransferase LgtC
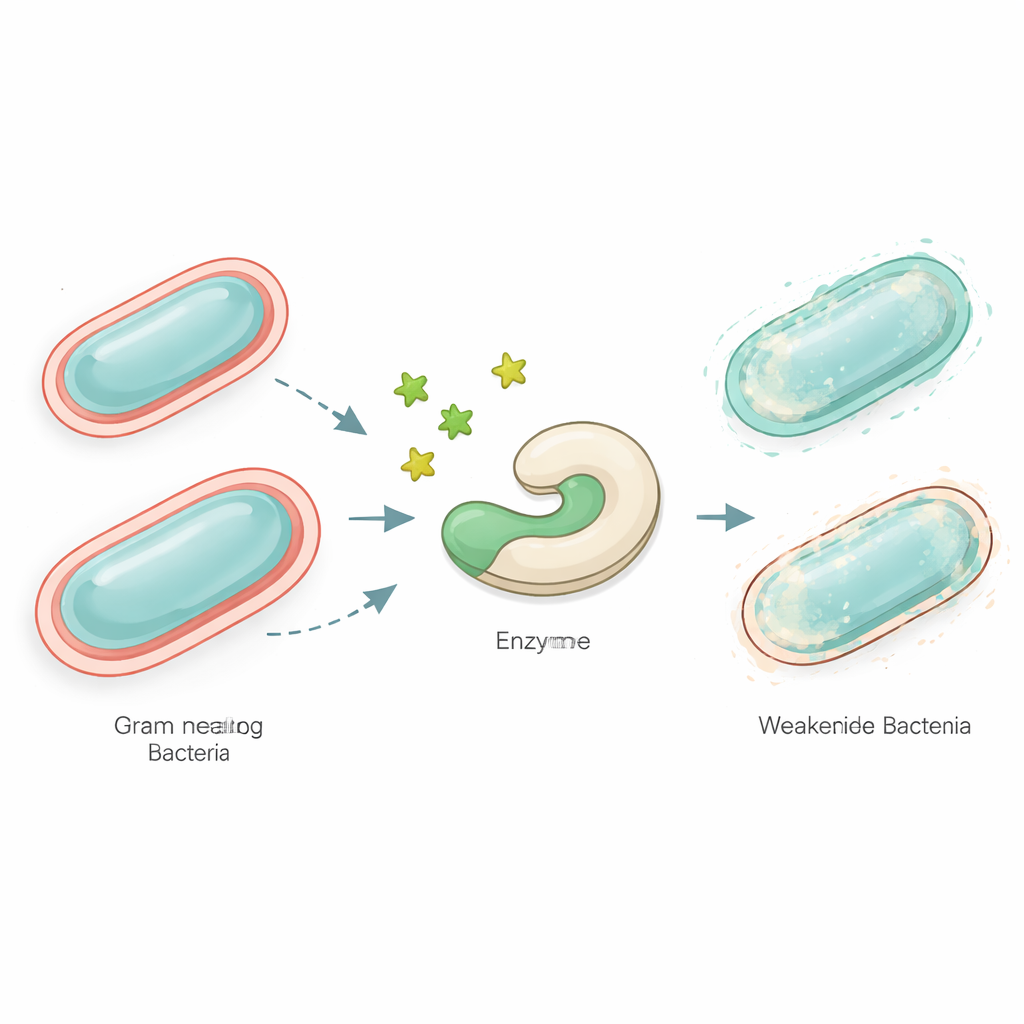
Why weakening germs can be better than killing them
Hospitals around the world are struggling with infections caused by bacteria that no longer respond to many antibiotics. Instead of trying to kill these microbes outright, some scientists are exploring a different tactic: quietly disarming them so our immune systems and existing drugs can do the rest. This study uses computer models to hunt for new small molecules, called thioglycosides, that could block a key bacterial enzyme and make dangerous Gram‑negative pathogens much less capable of causing disease.
The problem with tough outer shields
Many of the most worrisome bacteria, such as Neisseria species and other Gram‑negative germs, are wrapped in a complex outer coat of sugary molecules. Part of this coat, known as lipooligosaccharide, helps them evade our immune defenses and resist antibiotics. Building this shield requires a set of specialized enzymes. One of them, called LgtC, attaches a sugar called galactose to the growing outer layer. If LgtC is blocked, the surface structure is left incomplete, and the bacteria become more vulnerable. Because human cells do not use LgtC, it is an appealing target for drugs that might cripple pathogens without harming us.
Using computers to search chemical space
The researchers focused on a family of sugar‑like molecules called thioglycosides, which have a sulfur atom in place of an oxygen link found in natural sugars. Earlier work showed that two such compounds, named FucSBn and BacSBn, can interfere with bacterial sugar assembly. Here, the team searched the large PubChem database for molecules that closely resemble these two “metabolic decoys.” They kept only candidates that, based on standard drug‑likeness rules and predicted absorption and safety profiles, looked suitable for development as medicines. This filtering step yielded 18 promising analogs that were small enough, not too greasy or too water‑loving, and unlikely to be highly toxic at therapeutic doses.
Testing how well the candidates fit the enzyme
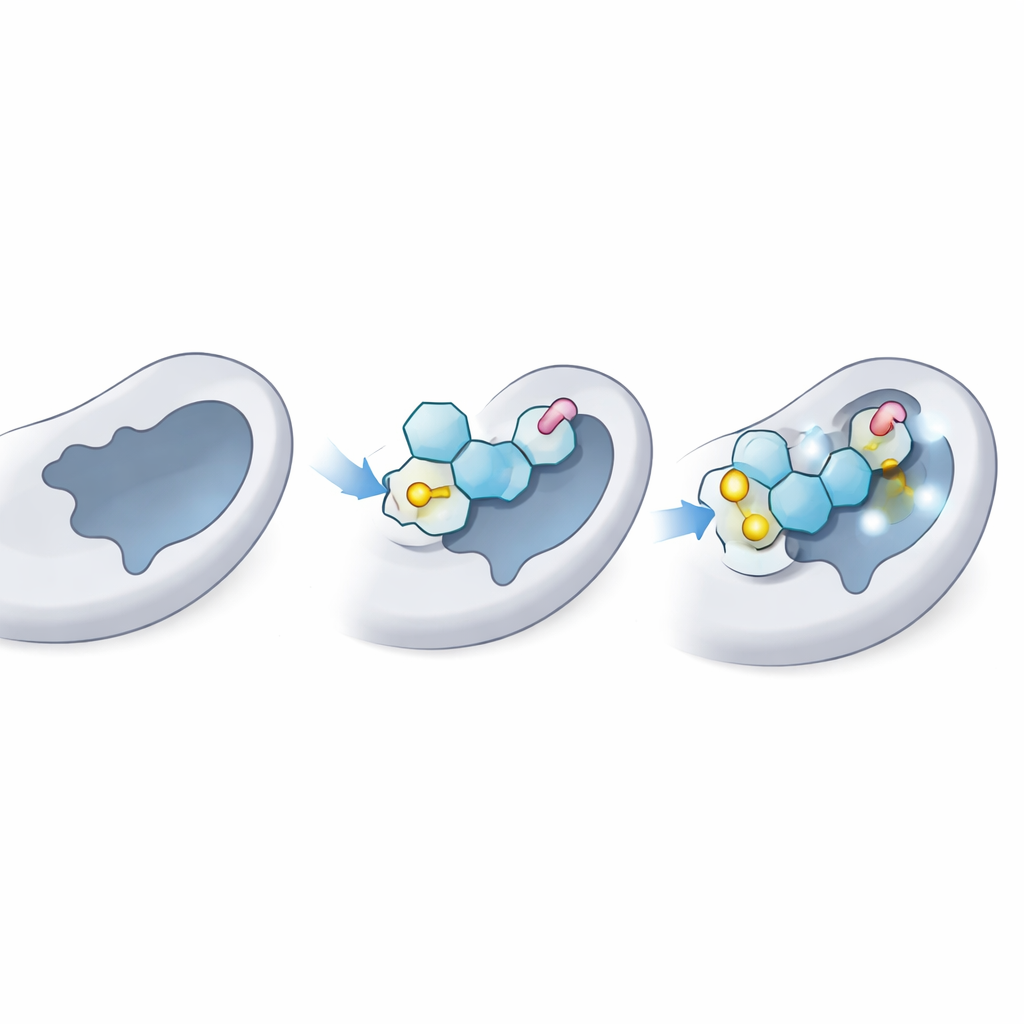
Next, the scientists used molecular docking, a kind of virtual lock‑and‑key test, to see how snugly each thioglycoside might sit in the donor site of LgtC—the pocket where the natural sugar donor normally binds. They first confirmed that their method could correctly “redock” the natural donor sugar into the known 3D structure of LgtC, matching experimental data. Then they docked all 18 analogs thousands of times. Several, especially three labeled C‑5, C‑14, and C‑18, consistently showed stronger predicted binding than the natural donor, suggesting that they could effectively compete for the same spot on the enzyme.
Watching the interaction in motion
Docking offers a still image; the team then ran 100‑nanosecond molecular dynamics simulations to see how the enzyme and each ligand behaved over time in a virtual watery environment. These simulations track how much the complex wobbles, how compact the protein remains, and which contacts persist. The best thioglycosides kept a stable pose in the donor pocket, with only modest motion comparable to the natural sugar. They maintained key hydrogen bonds and close‑range contacts with the same anchor residues that normally hold the real donor in place, while adding extra stabilizing interactions thanks to their sulfur linkages and aromatic “tails.” Overall protein shape and flexibility stayed within healthy limits, indicating that the enzyme was not being distorted, just blocked.
What this means for future treatments
Taken together, the virtual screening, docking scores, and long simulations point to a small set of thioglycoside scaffolds—especially C‑5, C‑14, and C‑18—as strong candidates to act as competitive blockers of LgtC. In simple terms, these molecules appear capable of sitting in the enzyme’s sugar‑binding site long enough and tightly enough to prevent the real building blocks from getting in. That should disrupt construction of the bacteria’s protective outer coat, weakening the microbes without necessarily killing them outright. The work is entirely computational and still needs to be confirmed in enzyme and cell experiments, but it offers a clear roadmap for chemists: these thioglycosides provide starting blueprints for designing next‑generation antivirulence drugs that could help tame multidrug‑resistant Gram‑negative infections.
Citation: Sierra-Hernández, O., Saurith-Coronell, O., Alcázar, J.J. et al. In silico discovery of thioglycoside analogues as donor-site inhibitors of glycosyltransferase LgtC. Sci Rep 16, 13807 (2026). https://doi.org/10.1038/s41598-026-43638-x
Keywords: antivirulence, Gram-negative bacteria, glycosyltransferase LgtC, thioglycoside inhibitors, in silico drug discovery