Clear Sky Science · es
Descubrimiento in silico de análogos tiofílicos como inhibidores del sitio donante de la glicosiltransferasa LgtC
Por qué debilitar a los microbios puede ser mejor que matarlos
Los hospitales de todo el mundo lidian con infecciones causadas por bacterias que ya no responden a muchos antibióticos. En lugar de intentar eliminar estos microbios de forma directa, algunos científicos exploran una táctica diferente: desarmarlos silenciosamente para que nuestro sistema inmunitario y los medicamentos existentes puedan hacer el resto. Este estudio usa modelos por ordenador para buscar nuevas moléculas pequeñas, llamadas tiofínidos, que podrían bloquear una enzima bacteriana clave y hacer que patógenos Gram‑negativos peligrosos sean mucho menos capaces de causar enfermedad.
El problema de los resistentes escudos exteriores
Muchas de las bacterias más preocupantes, como especies de Neisseria y otros microbios Gram‑negativos, están envueltas en una capa exterior compleja de moléculas azucaradas. Parte de esta cobertura, conocida como lipooligosacárido, les ayuda a evadir nuestras defensas inmunitarias y a resistir los antibióticos. Construir este escudo requiere un conjunto de enzimas especializadas. Una de ellas, llamada LgtC, une un azúcar llamado galactosa a la capa exterior en formación. Si se bloquea LgtC, la estructura superficial queda incompleta y las bacterias se vuelven más vulnerables. Dado que las células humanas no usan LgtC, es un objetivo atractivo para fármacos que podrían incapacitar a los patógenos sin dañarnos.
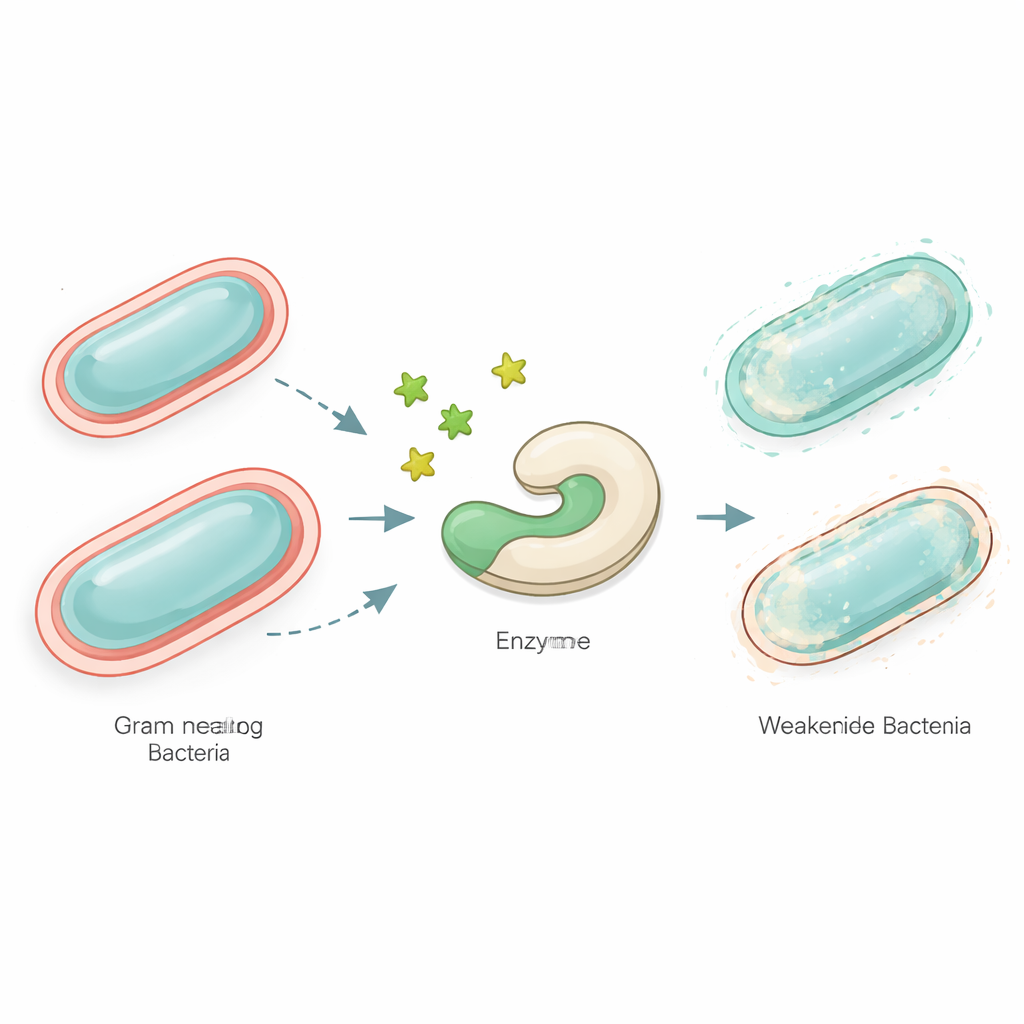
Usar ordenadores para explorar el espacio químico
Los investigadores se centraron en una familia de moléculas similares a azúcares llamadas tiofílicos, que tienen un átomo de azufre en lugar del enlace oxígeno presente en los azúcares naturales. Trabajos previos mostraron que dos de estos compuestos, denominados FucSBn y BacSBn, pueden interferir con el ensamblaje de azúcares bacterianos. Aquí, el equipo exploró la amplia base de datos PubChem en busca de moléculas que se parecieran estrechamente a estos dos “señuelos metabólicos”. Conservó solo los candidatos que, según las reglas estándar de semejanza a fármacos y los perfiles predichos de absorción y seguridad, parecían adecuados para su desarrollo como medicamentos. Este filtro produjo 18 análogos prometedores que eran lo suficientemente pequeños, ni demasiado lipofílicos ni demasiado hidrofílicos, y con baja probabilidad de ser altamente tóxicos en dosis terapéuticas.
Probar qué tal encajan los candidatos en la enzima
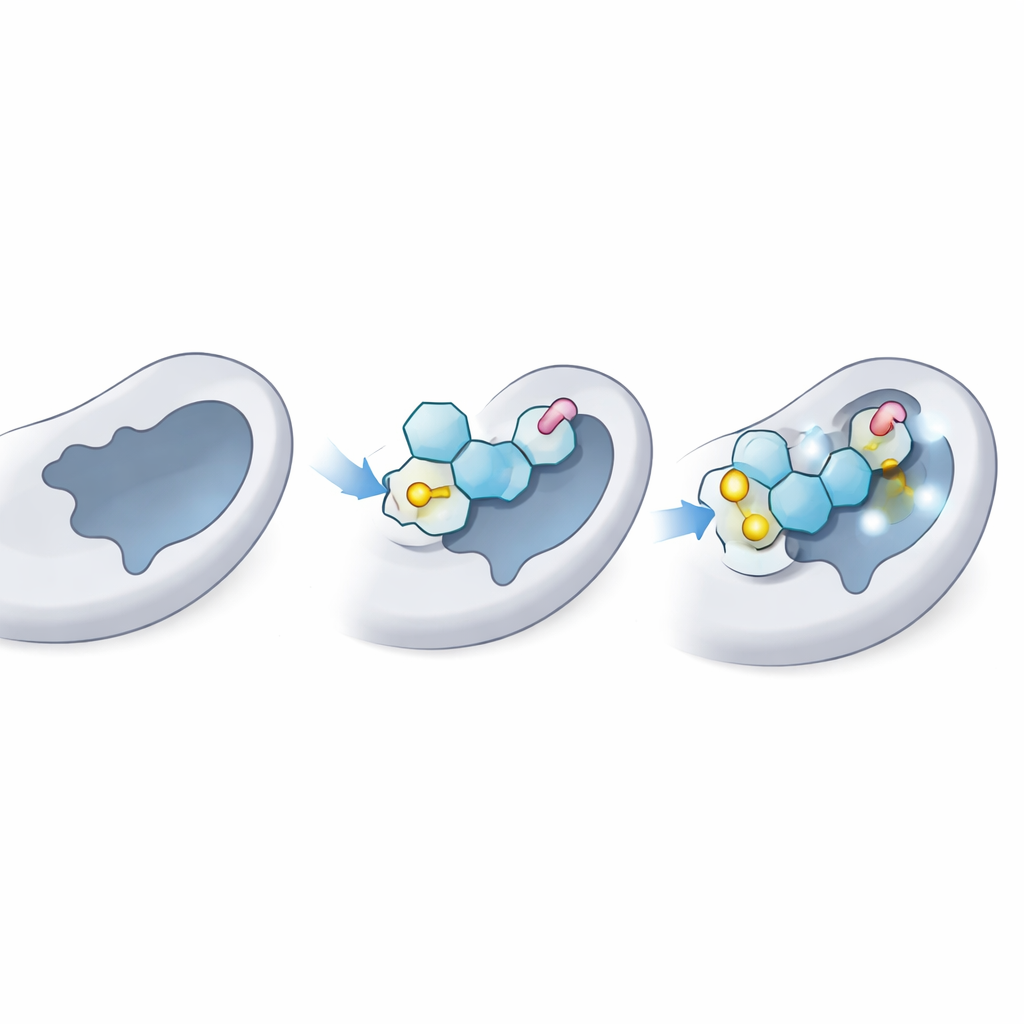
A continuación, los científicos utilizaron acoplamiento molecular, una especie de prueba virtual de cerradura y llave, para ver con qué ajuste podría sentarse cada tiofílico en el sitio donante de LgtC—el bolsillo donde normalmente se une el donador azucarado natural. Primero confirmaron que su método podía “reubicar” correctamente el azúcar donador natural en la estructura 3D conocida de LgtC, coincidiendo con datos experimentales. Luego acoplaron los 18 análogos miles de veces. Varios, especialmente tres etiquetados C‑5, C‑14 y C‑18, mostraron de forma consistente una afinidad predicha mayor que la del donador natural, lo que sugiere que podrían competir eficazmente por el mismo sitio en la enzima.
Observar la interacción en movimiento
El acoplamiento ofrece una imagen fija; el equipo ejecutó entonces simulaciones de dinámica molecular de 100 nanosegundos para ver cómo se comportaban la enzima y cada ligando a lo largo del tiempo en un entorno acuoso virtual. Estas simulaciones rastrean cuánto se tambalea el complejo, cuán compacto permanece la proteína y qué contactos persisten. Los mejores tiofílicos mantuvieron una pose estable en el bolsillo donante, con solo un movimiento modesto comparable al del azúcar natural. Mantuvieron enlaces de hidrógeno clave y contactos a corta distancia con los mismos residuos ancla que normalmente sostienen al donador real, a la vez que añadían interacciones estabilizadoras adicionales gracias a sus enlaces de azufre y colas aromáticas. La forma y flexibilidad global de la proteína se mantuvieron dentro de límites saludables, indicando que la enzima no se estaba distorsionando, solo bloqueándose.
Qué implica esto para tratamientos futuros
En conjunto, el cribado virtual, las puntuaciones de acoplamiento y las simulaciones prolongadas señalan a un pequeño conjunto de andamios tiofílicos—especialmente C‑5, C‑14 y C‑18—como candidatos sólidos para actuar como bloqueadores competitivos de LgtC. En términos sencillos, estas moléculas parecen capaces de ocupar el sitio de unión al azúcar de la enzima el tiempo suficiente y con la suficiente fuerza como para impedir que entren los bloques de construcción reales. Eso debería interrumpir la construcción de la capa protectora bacteriana, debilitando a los microbios sin necesariamente matarlos por completo. El trabajo es enteramente computacional y aún necesita confirmación en experimentos con enzima y en células, pero ofrece una hoja de ruta clara para los químicos: estos tiofílicos proporcionan planos iniciales para diseñar la próxima generación de fármacos antivirulencia que podrían ayudar a controlar infecciones Gram‑negativas multirresistentes.
Cita: Sierra-Hernández, O., Saurith-Coronell, O., Alcázar, J.J. et al. In silico discovery of thioglycoside analogues as donor-site inhibitors of glycosyltransferase LgtC. Sci Rep 16, 13807 (2026). https://doi.org/10.1038/s41598-026-43638-x
Palabras clave: antivirulencia, bacterias Gram‑negativas, glicosiltransferasa LgtC, inhibidores tiofílicos, descubrimiento de fármacos in silico