Clear Sky Science · pl
Odkrycie in silico analogów tioglikozydów jako inhibitorów miejsca donorowego glikozylotransferazy LgtC
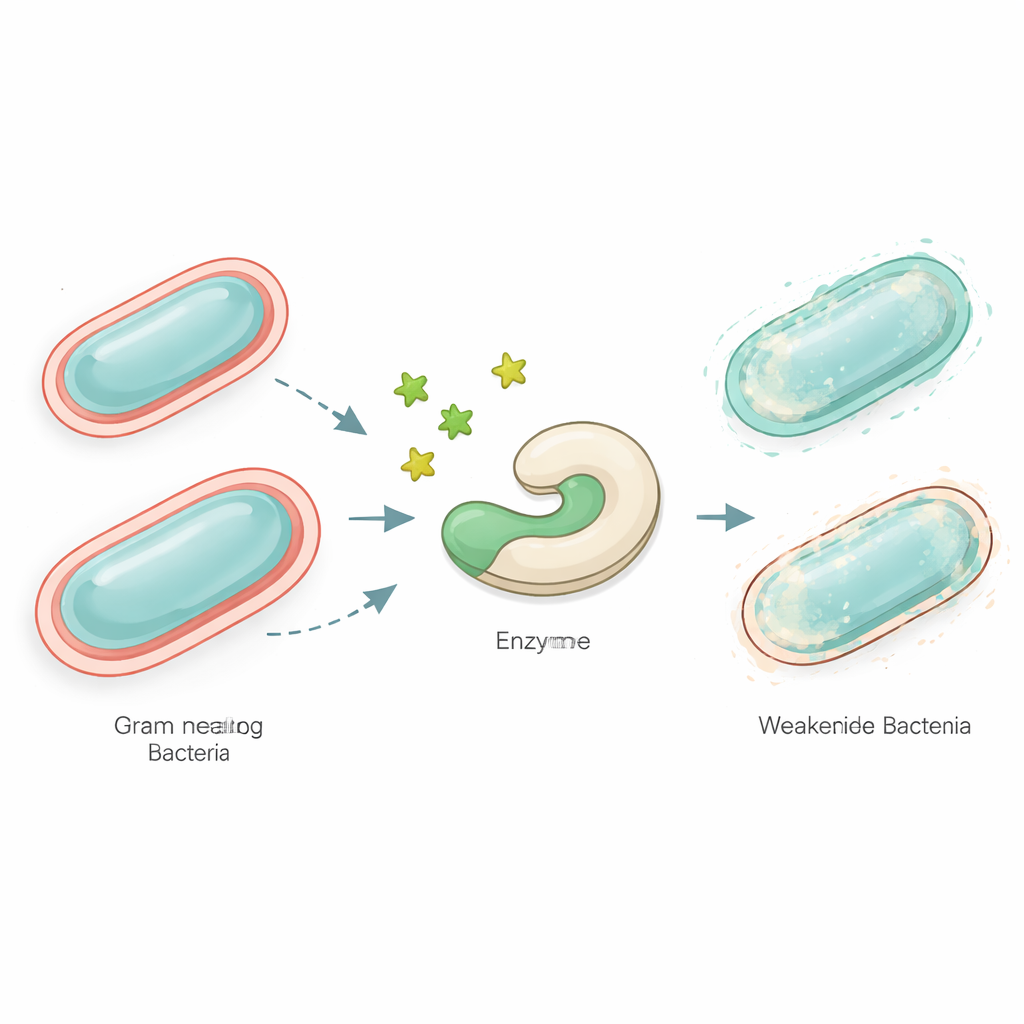
Dlaczego osłabianie zarazków może być lepsze niż ich zabijanie
Szpitale na całym świecie borykają się z zakażeniami wywołanymi przez bakterie, które przestały reagować na wiele antybiotyków. Zamiast próbować je natychmiastowo zabijać, niektórzy naukowcy badają inną taktykę: dyskretne rozbrajanie ich, by nasz układ odpornościowy i istniejące leki mogły dokończyć pracę. W tym badaniu wykorzystano modele komputerowe do poszukiwania nowych małych cząsteczek, zwanych tioglikozydami, które mogłyby blokować kluczowy enzym bakteryjny i znacznie ograniczyć zdolność niebezpiecznych patogenów Gram‑ujemnych do wywoływania chorób.
Problem z twardymi zewnętrznymi osłonami
Wiele z najbardziej niepokojących bakterii, takich jak gatunki Neisseria i inne drobnoustroje Gram‑ujemne, jest otoczonych złożoną zewnętrzną powłoką z cząsteczek cukrowych. Część tej powłoki, znana jako lipooligosacharyd, pomaga im unikać obrony immunologicznej i opierać się antybiotykom. Budowa tej osłony wymaga zestawu wyspecjalizowanych enzymów. Jeden z nich, nazwany LgtC, przyłącza cukier o nazwie galaktoza do powstającej warstwy zewnętrznej. Jeśli LgtC zostanie zablokowany, struktura powierzchni pozostaje niekompletna, a bakterie stają się bardziej podatne. Ponieważ komórki ludzkie nie używają LgtC, jest to atrakcyjny cel dla leków, które mogłyby unieruchomić patogeny bez szkody dla nas.
Wykorzystanie komputerów do przeszukiwania przestrzeni chemicznej
Naukowcy skupili się na rodzinie cząsteczek przypominających cukry, zwanych tioglikozydami, które mają atom siarki zamiast wiązania tlenowego występującego w naturalnych cukrach. Wcześniejsze badania wykazały, że dwa takie związki, nazwane FucSBn i BacSBn, mogą zakłócać montaż cukrowy bakterii. W tej pracy zespół przeszukał dużą bazę PubChem w poszukiwaniu cząsteczek ściśle przypominających te dwa „metaboliczne atrapy”. Zachowano tylko kandydatów, którzy na podstawie standardowych reguł „drug‑likeness” oraz przewidywanych profili wchłaniania i bezpieczeństwa wydawali się odpowiedni do rozwoju jako leki. Ten etap filtrowania przyniósł 18 obiecujących analogów, które były wystarczająco małe, niezbyt tłuste ani nadmiernie hydrofilowe i mało prawdopodobne, by były wysoce toksyczne w dawkach terapeutycznych.
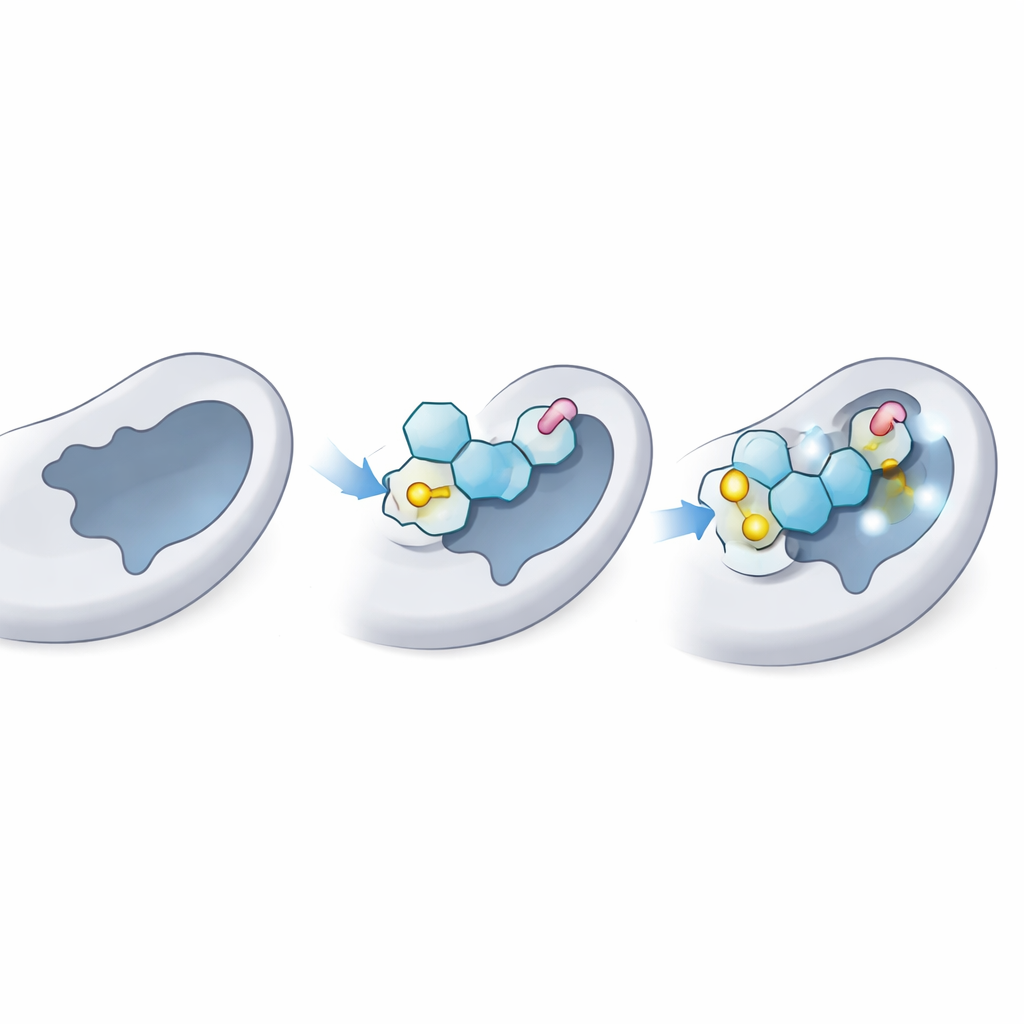
Badanie, jak dobrze kandydaci pasują do enzymu
Następnie naukowcy zastosowali dokowanie molekularne, rodzaj wirtualnego testu „zamek‑i‑klucz”, by sprawdzić, jak ciasno każdy tioglikozyd może siedzieć w miejscu donorowym LgtC — kieszonce, gdzie zwykle wiąże się naturalny dono r cukru. Najpierw potwierdzili, że ich metoda potrafi poprawnie „redockować” naturalny donor cukrowy do znanej struktury 3D LgtC, odpowiadając danym eksperymentalnym. Potem dokowali wszystkie 18 analogów tysiące razy. Kilka z nich, zwłaszcza trzy oznaczone C‑5, C‑14 i C‑18, konsekwentnie wykazywały silniejsze przewidywane wiązanie niż naturalny donor, co sugeruje, że mogłyby skutecznie konkurować o to samo miejsce na enzymie.
Obserwowanie interakcji w ruchu
Dokowanie daje obraz statyczny; zespół uruchomił więc symulacje dynamiki molekularnej trwające 100 nanosekund, aby zobaczyć, jak enzym i każdy ligand zachowują się z upływem czasu w wirtualnym wodnym środowisku. Symulacje śledzą, jak bardzo kompleks się chwieje, jak zwarty pozostaje białko i które kontakty utrzymują się. Najlepsze tioglikozydy utrzymywały stabilną pozycję w kieszonce donorowej, z jedynie umiarkowanym ruchem porównywalnym z naturalnym cukrem. Zachowywały kluczowe wiązania wodorowe i kontakty w bliskim zasięgu z tymi samymi resztami kotwiczącymi, które normalnie trzymają rzeczywistego donora, jednocześnie dodając dodatkowe stabilizujące interakcje dzięki swoim siarkowym wiązaniom i aromatycznym „ogonkom”. Ogólny kształt i elastyczność białka pozostały w zdrowych granicach, wskazując, że enzym nie był zdeformowany, lecz po prostu zablokowany.
Co to oznacza dla przyszłych terapii
Podsumowując, przegląd wirtualny, wyniki dokowania i długie symulacje wskazują na niewielki zestaw rusztowań tioglikozydów — szczególnie C‑5, C‑14 i C‑18 — jako mocnych kandydatów do działania jako inhibitory konkurencyjne LgtC. Mówiąc prościej, te cząsteczki wydają się zdolne do zajęcia miejsca wiążącego cukier w enzymie wystarczająco długo i na tyle mocno, by uniemożliwić wejście prawdziwych bloków budulcowych. To powinno zaburzyć budowę ochronnej zewnętrznej powłoki bakterii, osłabiając drobnoustroje bez konieczności ich bezpośredniego zabijania. Praca jest w całości obliczeniowa i wymaga potwierdzenia w eksperymentach z enzymami i komórkami, ale daje jasny plan dla chemików: te tioglikozydy stanowią punkty wyjścia do projektowania następnej generacji leków przeciwko wirulencji, które mogłyby pomóc poskromić wielolekooporne zakażenia Gram‑ujemne.
Cytowanie: Sierra-Hernández, O., Saurith-Coronell, O., Alcázar, J.J. et al. In silico discovery of thioglycoside analogues as donor-site inhibitors of glycosyltransferase LgtC. Sci Rep 16, 13807 (2026). https://doi.org/10.1038/s41598-026-43638-x
Słowa kluczowe: antywirotliwość, bakterie Gram-ujemne, glikozylotransferaza LgtC, inhibitory tioglikozydowe, odkrywanie leków in silico